 2022, Vol. 40
2022, Vol. 40Institute of Oceanology, Chinese Academy of Sciences
Article Information
- GONG Jie, SHEN Guoqing, ZHU Mengru, ZHAN Ming, XI Changjun, SHUI Yan, XU Zenghong, SHEN Huaishun
- 16S rRNA gene sequencing analysis on changes in the intestinal flora of Procambarus clarkii with "Black May" disease
- Journal of Oceanology and Limnology, 40(5): 2068-2079
- http://dx.doi.org/10.1007/s00343-021-1278-4
Article History
- Received Sep. 3, 2021
- accepted in principle Oct. 8, 2021
- accepted for publication Nov. 26, 2021




2 Key Laboratory of Freshwater Fisheries and Germplasm Resources Utilization, Ministry of Agriculture, Freshwater Fisheries Research Center, Chinese Academy of Fishery Sciences, Wuxi 214081, China
Convincing evidence suggests that the intestinal microbiota plays an important role in maintaining host health, including nutrient absorption, pathogen defense, and antibiotic resistance (Ott et al., 2004; Sekirov et al., 2010; Dai et al., 2017). In addition, the intestinal microbial flora, which produces signaling molecules, can influence host metabolism, such as short-chain fatty acid (SCFA) production and vitamin synthesis (Watanabe et al., 2006). Therefore, the balance of the intestinal ecosystem is very important for host health, and a change in bacterial diversity may lead to host disease (Guarner, 2007; Othman et al., 2008). The outbreak of diseases in aquaculture is usually accompanied by disorder of the intestinal flora. Therefore, characterizing the differences in intestinal flora between healthy and diseased hosts can be a first step in improving disease prevention and treatment (Berry et al., 2012; Berry and Reinisch, 2013). Studies have explored the differences in intestinal microflora between healthy and diseased aquatic animals. In Penaeus vannamei with acute hepatopancreatic necrosis disease (AHPND), Vibrio was enriched in the intestine (Dong et al., 2021). There were significant differences in intestinal microflora between healthy and diseased (suffering from furunculosis) Coreius guichenoti (Li et al., 2016). Some studies have shown that changes in the intestinal bacterial community are closely related to the severity of P. vannamei, and the indicator group can be used for health assessment (Xiong et al., 2015). However, the analysis of crustacean intestinal microbiota is still in its infancy, and it is not clear how diseases affect the intestinal microbiota.
Procambarus clarkii, a species of crayfish, is classified into arthropods, crustaceans, Decapoda, and the family Labridae. P. clarkii is native to northern Mexico and the southern USA, and it has spread to Europe, Africa, Asia, and other parts of the Americas (Gherardi, 2006). The species was introduced to China in 1929 (Yue et al., 2008). In recent years, it has become an important species in the aquaculture industry due to its high protein, rich nutrition, and delicious flavor. However, the peak of the disease season typically occurs in May, during the farming of crayfish, and the phenomenon occurred in May is commonly known as "Black May" disease (BMD) (Huang et al., 2020). Diseased crayfish usually exhibit reduced food intake. Their carapace is easily peeled off and movement is slow. Dissection of diseased individuals reveals that the intestine is empty, blue, and swollen (Fig. 1a). BMD has caused tremendous economic losses to the crayfish farming industry. Once a pond becomes affected, nearly 90% mortality would be typically reported. At present, there are few studies on BMD in P. clarkii in China, so little is known about how BMD affects P. clarkii. Considering that the intestines of P. clarkii with BMD are empty and swollen, it is necessary to characterize the changes in intestinal microbiota.

|
| Fig.1 The intestines of diseased (a) and healthy (b) P. clarkii The intestines of healthy P. clarkii was black and full, and the intestines of diseased P. clarkii was blue, empty, and swollen. |
To explore the influence of BMD on the intestinal flora of P. clarkii, we compared the intestinal flora of healthy and BMD-diseased P. clarkii, and found associated changes in the intestinal flora. The results provide a theoretical reference for the study of the intestinal bacteria of P. clarkii and will help in the prevention and treatment of this disease.
2 MATERIAL AND METHOD 2.1 SamplingThe diseased crayfish used in the experiment were collected from a P. clarkii farm with BMD in Xuyi County, Jiangsu Province, China. The organisms exhibited reduced food intake and lethargy. Dissection of diseased individuals reveals that the intestine is empty, blue, and swollen. Our samples (control and diseased) were collected from different ponds. The intestines of 10 healthy (control) and 10 diseased P. clarkii were collected, frozen in liquid nitrogen, and stored at -80 ℃. And our laboratory through PCR amplification and observation with transmission electron microscopy to detect the pathogens in the sampled crayfish, including white spot syndrome virus (WSSV) and Procambarus clarkii dicistro-like virus (PcDV, a novel Dicistro-like virus discovered in Procambarus clarkii with "Black May"disease). In the end, both pathogens were positive (Huang et al., 2020).
2.2 Extraction of genomic DNA and PCR amplification of 16S rDNAThe total genomic DNA of P. clarkii intestine was extracted by the cetyltrimethylammonium bromide (CTAB) method, and the concentration of isolated DNA was measured with a NanoDrop 2000 spectrophotometer (GE Healthcare, USA). The samples were diluted to 1 ng/μL with sterile water and used to amplify the 16S rDNA of intestinal microorganisms.
The diluted genomic DNA was used as the template for PCR amplification. PCR amplification of the 16S rRNA gene was performed using PCR primers (515F-806R) specific for the V3–V4 regions. The primer sequences were as follows: 5ʹ-GTGCCAGCMGCC-GCGGTAA-3ʹ/5ʹ-GGACTACHVGGGTWTCTAAT-3ʹ. PCR products were detected by electrophoresis with 2% agarose gels. Then, PCR products were purified with GeneJET Gel Extraction Kit (Thermo Scientific). Before sequencing, a spectrophotometer was used to determine the DNA concentration of each PCR product, and the DNA was mixed in an appropriate ratio according to the sequencing requirements.
2.3 High-throughput sequencingTwenty samples were sequenced in a commercial company (Novogene, Beijing), of which 10 samples were from the control group and 10 were from the diseased group. Sequencing libraries were generated using an Illumina TruSeq DNA PCR-Free Library Preparation Kit (Illumina, USA) following the manufacturer's recommendations and index adapters were added. The library quality was assessed on the Qubit 2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system. Finally, the library was sequenced on an Illumina Nova Seq platform and 250-bp paired-end reads were generated.
2.4 Sequence analysisPaired-end reads were assigned to samples based on their unique barcode and truncated by cutting off the barcode and primer sequence. Paired-end reads were merged using FLASH (V1.2.7, http://ccb.jhu.edu/software/FLASH/) to obtain the Raw tags (the spliced tags sequence) (Magoč and Salzberg, 2011). According to the tag quality control process of Qiime (V1.9.1, http://qiime.org/scripts/split_libraries_fastq.html) (Caporaso et al., 2010), the following operations were carried out: tags interception: truncate raw tags from the first low-quality base site with continuous low-quality value (the default quality threshold is ≤19) and the base number reaches the set length (the default length is 3); tags length filtering: filter out tags with continuous high-quality base length less than 75% of tags length. Effective tags were obtained through read splicing, quality control, and removal of chimeric sequences (https://github.com/torognes/vsearch/) (Rognes et al., 2016).
2.5 Statistical methodTo study the species composition of each sample, the effective tags of all samples were clustered into operational taxonomic units (OTUs) with 97% identity (Uparse v7.0.1001, http://www.drive5.com/uparse/) (Haas et al., 2011). The OTU sequences were taxonomically annotated by Mothur and SILVA132 (http://www.arb-silva.de/). According to the OTU clustering results, a Venn diagram was drawn to show common and unique OTUs between different groups (http://bioinformatics.psb.ugent.be/webtools/Venn/). According to the species annotation results, the relative abundances of species at different taxonomic levels were plotted.
The evaluation of microbial diversity in a natural environment involves two aspects: species richness and evenness information. Alpha diversity was used to analyze the richness and diversity of the within-community microbial community, as shown by rank abundance. Alpha diversity indices (Good's coverage, the Chao1 estimator, the ACE, the Simpson index, and the Shannon index) were calculated in QIIME (Version 1.9.1) to analyze the abundance and diversity of microbial communities in the two groups. R (version 2.15.3) was used to analyze the difference of alpha diversity index between groups. Beta diversity was used to analyze the microbial community composition of different samples. QIIME was used to calculate Unweighted UniFrac distance and construct unweighted pair group method with arithmetic (UPGMA) sample clustering tree. Principal coordinate analysis (PCoA) was conducted by the WGCNA packages, STAT packages, and GGPLOT2 packages of R (Version 2.15.3), based on Unweighted UniFrac distance. Moreover, linear discriminant analysis (LDA) effect size (LEfSe) was used to identify biomarkers with significant differences between the groups (the default filter value of the LDA score is 4).
The 16S rRNA gene sequences of prokaryotes in the kyoto encyclopedia of genes and genomes (KEGG) database were extracted and compared to those in the SILVA SSU Ref NR database (BLAST bitcore > 1 500) by the BLASTN algorithm to establish the correlation matrix. The KEGG database prokaryotic whole genome function information annotated by UProC and PAUDA was mapped to the SILVA database to realize the functional annotation of the SILVA database.
3 RESULT 3.1 The high-throughput sequencing data analysisIn total, through the splicing of reads, 85 772 tags per sample were measured, and 82 002 valid data were obtained through quality control, with the amount of effective data reaching 77 703 tags after removing chimaera sequences. With 97% identity, the sequences were grouped into OTUs, with 2 432 OTUs. In the Venn diagram, the two circles represent the diseased group and the control group, and the numbers represent the number of OTUs. Among them, 1 260 OTUs were shared between the two groups, 438 OTUs were unique to the diseased group, and 716 OTUs were unique to the control group (Fig. 2).

|
| Fig.2 The Venn diagrams (a) and the number of OTUs in control and diseased group (b) The shared and unique OTUs are represented through Venn diagrams; 1 260 OTUs are shared among samples. |
In this study, 47 phyla were recognized in the intestinal flora of control and diseased groups. As shown in Fig. 3, we identified four dominant phyla in the intestines of crayfish in the control group, namely Tenericutes (30.86%), Bacteroidetes (29.99%), Firmicutes (22.23%), and Proteobacteria (15.23%). We also identified three dominant phyla in the diseased group, namely, Tenericutes (35.71%), Proteobacteria (34.59%), and Firmicutes (26.65%). Although Tenericutes had the highest abundance in both groups, and there was no significant difference (P > 0.05), a striking shift in the microbial composition was found in the intestines of P. clarkii with BMD. Bacteroidetes was a dominant phylum in healthy P. clarkii, whereas its prevalence was low in diseased P. clarkii (1.87%). By contrast, the prevalence of Proteobacteria was significantly higher (P < 0.05) in P. clarkii with BMD than in P. clarkii without BMD. The percentage of bacteria belonging to the phylum Proteobacteria in diseased crayfish was higher than that in healthy crayfish, which was mainly due to the contributions of Alphaproteobacteria (P < 0.05). In contrast, the percentage of Bacteroides in diseased crayfish was significantly lower than that in healthy crayfish (P < 0.05).

|
| Fig.3 Abundance of intestinal microflora of P. clarkii at the phylum level in the control and diseased groups *: P≤0.05; **: P≤0.01. |
As shown in Fig. 4a, the composition of the intestinal flora of P. clarkii showed more relative differences at the genus level. In the annotation results, a total of 1 150 (47.29%) OTUs annotations were at the genus level. Among them, the dominant intestinal bacteria of P. clarkii in the control group were Candidatus Bacilloplasma (47.56%) and Citrobacter (20.30%). The dominant intestinal bacteria in the diseased group were Candidatus Bacilloplasma (28.72%), Citrobacter (13.30%), and Aeromonas (20.76%). Although Candidatus Bacilloplasma was the dominant intestinal bacterium in both the control group and the diseased group, its abundance was significantly lower (P < 0.01) in the diseasedd group. In addition, other genera with significant enrichment in the diseased group were Aeromonas (P < 0.01) and Vibrio (P < 0.01) (Fig. 4c). The individual relative abundances for samples in the control and diseased group were shown in Fig. 4b, and the detailed data were shown in Supplementary Tables S1 & S2.

|
| Fig.4 The relative abundances of the intestinal bacterial community among the diseased and control samples at the genus level The top 10 most abundant genera are shown. Other minor taxa and unclassified bacteria are listed under "Other". Means for each group (a); means for each sample (b); bacteria with significant differences (c). D1–D10: diseased group; C11–C1: control group. **: P≤0.01. |
The results of alpha diversity analysis show that the Simpson, ACE, Chao1, and Shannon indices in the diseased group were slightly lower than those in the control group (Supplementary Table S3), but there was no significant difference in alpha diversity indices between the groups (richness P=0.59; evenness P=0.43; and diversity P=0.052) (Fig. 5a).

|
| Fig.5 Measures of α-biodiversity, including richness (Chao1), evenness (Shannon), and diversity (Simpson) (a), and principal component analysis (PCA) (b) In (b), the first two PCs are used as the x and y coordinates. Each sample is represented as a point. Samples from different sources are colored differently: diseased (red); control (blue). |
PCoA of all 20 samples showed that the clustering of sample points in the control group and the diseased group were relatively close, indicating that the difference in intestinal flora among individuals within each group was small. In contrast, the clusters of sample points of the control group and the diseased group were far apart, indicating a certain difference between intestinal flora groups (Fig. 5b). PCoA indicated that PC1, which reflected the bacterial community, could separate the healthy group from the diseased group, and the total contribution of other principal components was 15.08%.
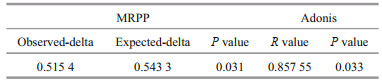
In addition, a multi response permutation procedure (MRPP, based on Bray-Curtis distance) analysis was performed to examine the differences in intestinal microbiota between the two groups, as shown in Table 1. The difference between groups was expressed as the expected-delta, which was greater than the difference within groups (significance=0.031).
|
To further analyze the intestinal flora of P. clarkii related to BMD, LEfSe was used to compare the relative abundance of intestinal flora between the control group and the diseased group (Fig. 6a). There were 5 significantly different flora constituents between the control group and the diseased group, belonging to three phyla: Cyanobacteria, Firmicutes, and Proteobacteria. Among them, we found that the bacteria of the diseased group showed a significant increase in the abundances of Enterobacteriaceae and Vibrionaceae of Proteobacteria. Their relationship is shown in Fig. 6b.

|
| Fig.6 Linear discriminant analysis (LDA) (a) and Cladogram analysis (b) The histogram of LDA value distribution shows species with LDA score greater than the set value (the default setting is 4), and the length of the histogram represents the impact of different species. In this branching diagram of evolution, the concentric circles from the inside to the outside represents the taxonomic rank from phylum to genus (or species). Each node represents a classification at that level, and the diameter of the node is proportional to the relative abundance. |
To analyze the functional changes in intestinal microbiota in P. clarkii with BMD, Tax4Fun was used to predict the metagenomic potential between the two groups. The results show that the high abundance of bacterial metagenomes in the intestine of healthy and BMD-diseased P. clarkii was mainly related to the metabolic pathway group, genetic information processing group and environmental information processing group at KEGG level 1. There was no significant difference between the KEGG level 1 microbial function of the diseased group and the control group (P > 0.05). For the metabolism group, the largest abundance was found in amino acid metabolism, carbohydrate metabolism, and energy metabolism in level 2. For the genetic information processing group, the highest abundance was found in replication and repair and translation. In the environmental information processing group, the greatest abundance was observed in the membrane transport pathway. The specific data analysis of the above results is shown in Supplementary Fig.S1. Specifically, the microbial functions related to replication and repair in diseased group were significantly more abundant (P < 0.05) than those in the control group. More detailed changes were observed at KEGG Level 2. As shown in Fig. 7, the microbial functions related to Glycan biosynthesis and metabolism and Enzyme families in the diseased group were significantly more enriched (P < 0.05) than those in the control group, while those assigned to the Amino acid metabolism, Energy metabolism, and Metabolism of cofactors and vitamins were significantly more enriched (P < 0.05) in the control group than in the diseased group.

|
| Fig.7 Extended error bar chart to determine the significant difference (95% confidence interval) between the average proportion of functional genes in the control (blue) and diseased (orange) samples The corrected P value is displayed on the right side. |
Procambarus clarkii is widely cultivated in China because of its strong environmental adaptability and great commercial value. As the scale of production has increased, outbreaks of disease in P. clarkii have occurred frequently, especially around May, when the incidence rate and mortality rate of P. clarkii reach their peak, phenomenon known as "Black May" disease (BMD). At present, there are many controversies about the cause of BMD. It is generally considered that BMD in P. clarkii is caused by large fluctuations in temperature and water temperature in May. WSSV and some Vibrio species reproduce in large numbers and cause proliferative diseases in P. clarkii (Chen, 2019). BMD is also believed to be caused by the accumulation of ammonia nitrogen and nitrite due to the rapid rise of water temperature and low dissolved oxygen in the water (Shen et al., 2020). According to the symptoms of the empty, swollen intestines of P. clarkii with BMD, the characteristics and structure of intestinal bacteria in crayfish with BMD and healthy crayfish were determined by 16S rRNA gene sequencing.
Crustaceans, such as P. clarkii, have a large number of microorganisms inhabiting their intestines. These microorganisms are the products of long-term coevolution with the host (Serrano-Villar et al., 2016). We identified four dominant phyla, namely, Proteobacteria, Bacteroidetes, Firmicutes, and Tenericutes, in the intestines of healthy crayfish, which confirms a previous study on the dominance of these taxa in the intestinal bacterial community of P. clarkii (Liu et al., 2020a; Zhang et al., 2020, 2021). In the present study, the alpha diversity of the bacteria in the intestinal tract of P. clarkii did not differ significantly (P > 0.05) between control and diseased groups. However, there was a significant difference in the prevalence of the main bacterial phyla in the intestine of the groups with and without BMD, which changed the composition of the intestinal flora. We found that compared with healthy crayfish, there was a significant enrichment of Proteobacteria (P < 0.05) in the intestines of diseased crayfish (Fig. 3). In contrast, Bacteroides was prevalent in the intestines of healthy crayfish, while its prevalence was low in diseased crayfish. As the most abundant bacteria in fishponds, Proteobacteria participates in various biogeochemical processes in aquatic ecosystems (such as carbon, nitrogen, and sulfur cycles) (Klase et al., 2019; Li et al., 2021). Studies have shown that Proteobacteria may be essential for certain physiological and biochemical functions of crustacean intestines, such as Portunus trituberculatus (Zeng et al., 2016; Li et al., 2018). And an increased abundance of Proteobacteria was a potential risk of disease in L. vannamei (Fan and Li, 2019). Bacteroides may be related to intestinal fat storage, and the nutrient absorption of Bacteroides is negatively correlated with the presence of fat deposits (Xu et al., 2007; Guo et al., 2008). The balance of the intestinal microbial population is very important to the health of the host, and a loss or increase in intestinal microbial diversity can lead to host diseases (Semova et al., 2012; Li et al., 2021). The enrichment of Proteobacteria abundance and the decrease in Bacteroides indicate that BMD may affect the fat metabolism and nutrient absorption function of the intestinal microbiota of P. clarkii. In addition, it was reported that Alphaproteobacteria may cause local infection of tissue in Litopenaeus vannamei(Huang et al., 2016). Alphaproteobacteria are enriched in the intestines of diseased crayfish (P < 0.05). However, further studies are needed to determine whether Alphaproteobacteria is related to damage to the intestinal tissues of P. clarkii with BMD.
At the genus level, the difference in the abundance of the intestinal flora of P. clarkii between the control group and the diseased group was mainly observed in Candidatus Bacilloplasma (P < 0.01), Bacteroides (P < 0.01), Aeromonas (P < 0.01) and Vibrio (P < 0.01) (Fig. 4c). Candidatus Bacilloplasma is considered to be a novel lineage of Mollicutes associated with the hindgut wall of the terrestrial isopod Porcellio scaber (Crustacea: Isopoda) and was first discovered in Porcellio scaber (Kostanjšek et al., 2007). The enrichment of Candidatus Bacilloplasma was also previously found in the intestine of P. vannamei (Hou et al., 2018), suggesting that Candidatus Bacilloplasma is prevalent in the intestines of crustaceans. Members of the genus Bacteroides are necessary to break down complex molecules in the intestines and help the body's immune system fight against potentially harmful pathogens (Gregory et al., 2015). In addition, Bacteroides is important for maintaining intestinal homeostasis, especially through the production of short-chain fatty acids (SCFA) (Zhong et al., 2015). Moreover, studies have shown that Bacteroides is rich in enzymes related to carbohydrate transport and protein metabolism, which are essential processes of digestion (Karlsson et al., 2011; Zhou and Zhi, 2016). The over-enrichment of Bacteroides may be related to the regulation of the homeostasis of the intestinal flora by P. clarkii. The Tax4Fun function prediction further supports the view that the abundance of genes involved in metabolism (Energy metabolism and Metabolism of cofactors and vitamans) in the intestinal flora of P. clarkii with BMD is significantly lower (P < 0.05) than that of control P. clarkii (Fig. 7), which indicates that energy metabolism is disrupted by BMD. This also further explains the symptoms of the jejunum and reduced food intake of P. clarkii with BMD.
In the intestine of P. clarkii in BMD, we detected significant upregulation of some opportunistic pathogens: Vibrio (0.152%–4.939%, P < 0.01) and Aeromonas (from 3.965% to 20.758%, P < 0.01) (Fig. 4c). Generally, many members of the Vibrio genus are considered the main pathogens causing disease and death in aquatic animals. For example, Vibrio parahaemolyticus can cause AHPND and "gill rot disease" in shrimp and crayfish (Alloul et al., 2021). Studies on P. clarkii show that pathogenic bacteria such as Vibrio may be an opportunistic factor for disease (Shui et al., 2020). When the host's immunity decreases or the number of bacteria exceeds the normal value, host disease can occur (Hou et al., 2018). Aeromonas hydrophila, a more common conditional pathogen, exists widely in aquatic environments. Wang et al. (2004) noted that pathogenic Aeromonas hydrophila can cause many diseases, such as tremor and oedema, in Eriocheir sinensis, and it is also one of the main bacterial pathogens in shrimp and crab farming operation. Although conditional pathogenic bacteria are significantly enriched in the intestine of crayfish with BMD, further studies are needed to determine whether the two are related to the pathogenesis of BMD in crayfish.
In the present research, the functional prediction of the intestinal flora indicated that the metagenomic potential of the intestinal flora of P. clarkii has a higher abundance in the Carbohydrates metabolism and Amino and acids, which may be attributed to the carbohydrates and proteins from the host being the main energy-related nutrients for bacterial colonization (El Asmar et al., 2000; Shi and Walker, 2004; Hou et al., 2018). In addition, some important pathways related to Cellular processes, Environmental information processing, and Human disease functions also changed, indicating that BMD may affect the biochemical processes of P. clarkii intestinal bacteria at the cellular and molecular levels.
5 CONCLUSIONOverall, our study demonstrated that BMD causes changes in the intestinal flora of P. clarkii. Specifically, we found that the prevalence of bacteria belonging to the phylum Proteobacteria in the intestine of P. clarkii increased, while the prevalence of bacteria belonging to the phylum Bacteroidetes decreased, reflecting the abnormal metabolism and nutrient absorption of crayfish with BMD, which provides new insights for further research on the pathogenesis of BMD.
6 DATA AVAILABILITY STATEMENTAll raw data were submitted to the NCBI Sequence Read Archive (SRA) under accession number PRJNA692534. The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Electronic supplementary material
Supplementary material (Supplementary Tables S1–S3 and Supplementary Fig.S1) is available in the online version of this article at https://doi.org/10.1007/s00343-021-1278-4.
Alloul A, Wille M, Lucenti P, Bossier P, Van Stappen G, Vlaeminck S E. 2021. Purple bacteria as added-value protein ingredient in shrimp feed: Penaeus vannamei growth performance, and tolerance against Vibrio and ammonia stress. Aquaculture, 530: 735788.
DOI:10.1016/j.aquaculture.2020.735788 |
Berry D, Reinisch W. 2013. Intestinal microbiota: a source of novel biomarkers in inflammatory bowel diseases?. Best Practice & Research Clinical Gastroenterology, 27(1): 47-58.
DOI:10.1016/j.bpg.2013.03.005 |
Berry D, Schwab C, Milinovich G, Reichert J, Ben Mahfoudh K, Decker T, Engel M, Hai B, Hainzl E, Heider S, Kenner L, Müller M, Rauch I, Strobl B, Wagner M, Schleper C, Urich T, Loy A. 2012. Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. The ISME Journal, 6(11): 2091-2106.
DOI:10.1038/ismej.2012.39 |
Caporaso J G, Kuczynski J, Stombaugh J, Bittinger K, Bushman F D, Costello E K, Fierer N, Pena A G, Goodrich J K, Gordon J I, Huttley G A, Kelley S T, Knights D, Koenig J E, Ley R E, Lozupone C A, McDonald D, Muegge B D, Pirrung M, Reeder J, Sevinsky J R, Turnbaugh P J, Walters W A, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7(5): 335-336.
DOI:10.1038/nmeth.f.303 |
Chen X H. 2019. The reasons for the formation of "Black May" and prevention and control measures in rice field shrimp farming. Fishery Guide to be Rich, 16: 52-54.
|
Dai W F, Yu W N, Zhang J J, Zhu J Y, Tao Z, Xiong J B. 2017. The gut eukaryotic microbiota influences the growth performance among cohabitating shrimp. Applied Microbiology and Biotechnology, 101(16): 6447-6457.
DOI:10.1007/s00253-017-8388-0 |
Dong P S, Guo H P, Wang Y T, Wang R Y, Chen H P, Zhao Y J, Wang K, Zhang D M. 2021. Gastrointestinal microbiota imbalance is triggered by the enrichment of Vibrio in subadult Litopenaeusvannamei with acute hepatopancreatic necrosis disease. Aquaculture, 533: 736199.
DOI:10.1016/j.aquaculture.2020.736199 |
El Asmar R, Fasano A, Bamford P, Berti I, Not T, Catassi C, Coppa G V, Panigrahi P. 2000. Probiotics prevent zonulin-mediated intestinal barrier dysfunction secondary to bacterial colonization. Gastroenterology, 118(4): A815.
|
Fan L F, Li Q X. 2019. Characteristics of intestinal microbiota in the Pacific white shrimp Litopenaeus vannamei differing growth performances in the marine cultured environment. Aquaculture, 505: 450-461.
DOI:10.1016/j.aquaculture.2019.02.075 |
Gherardi F. 2006. Crayfish invading Europe: the case study of Procambarus clarkii. Marine and Freshwater Behaviour and Physiology, 39(3): 175-191.
DOI:10.1080/10236240600869702 |
Gregory K E, LaPlante R D, Shan G, Kumar D V, Gregas M. 2015. Mode of birth influences preterm infant intestinal colonization with Bacteroides over the early neonatal period. Advances in Neonatal Care, 15(6): 386-393.
DOI:10.1097/ANC.0000000000000237 |
Guarner F. 2007. Role of intestinal flora in health and disease. Nutrición Hospitalaria, 22(S2): 14-19.
|
Guo X, Xia X, Tang R, Zhou J, Zhao H, Wang K. 2008. Development of a real-time PCR method for Firmicutes and Bacteroidetes in faeces and its application to quantify intestinal population of obese and lean pigs. Letters in Applied Microbiology, 47(5): 367-373.
DOI:10.1111/j.1472-765X.2008.02408.x |
Haas B J, Gevers D, Earl A M, Feldgarden M, Ward D V, Giannoukos G, Ciulla D, Tabbaa D, Highlander S K, Sodergren E, Methe B, DeSantis T Z, The Human Microbiome Consortium, Petrosino J F, Knight R, Birren B W. 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Research, 21(3): 494-504.
DOI:10.1101/gr.112730.110 |
Hou D W, Huang Z J, Zeng S Z, Liu J, Wei D D, Deng X S, Weng S P, Yan Q Y, He J G. 2018. Intestinal bacterial signatures of white feces syndrome in shrimp. Applied Microbiology and Biotechnology, 102(8): 3701-3709.
DOI:10.1007/s00253-018-8855-2 |
Huang P D, Shen G Q, Gong J, Zhu M R, Wang Y, Zhang X, Hashimu Ame K, Zang Y N, Shen H S. 2020. A novel Dicistro-like virus discovered in Procambarus clarkii with "Black May" disease. Journal of Fish Diseases, 44(6): 803-811.
DOI:10.1111/jfd.13309 |
Huang Z B, Li X Y, Wang L P, Shao Z Z. 2016. Changes in the intestinal bacterial community during the growth of white shrimp, Litopenaeus vannamei. Aquaculture Research, 47(6): 1737-1746.
DOI:10.1111/are.12628 |
Karlsson F H, Ussery D W, Nielsen J, Nookaew I. 2011. A closer look at Bacteroides: phylogenetic relationship and genomic implications of a life in the human gut. Microbial Ecology, 61(3): 473-485.
DOI:10.1007/s00248-010-9796-1 |
Klase G, Lee S, Liang S, Kim J, Zo Y G, Lee J. 2019. The microbiome and antibiotic resistance in integrated fishfarm water: Implications of environmental public health. Science of the Total Environment, 649: 1491-1501.
DOI:10.1016/j.scitotenv.2018.08.288 |
Kostanjšek R, Štrus J, Avguštin G. 2007. "Candidatus Bacilloplasma, " a novel lineage of Mollicutes associated with the hindgut wall of the terrestrial isopod Porcellio scaber (Crustacea: Isopoda). Applied and Environmental Microbiology, 73(17): 5566-5573.
DOI:10.1128/AEM.02468-06 |
Li E C, Xu C, Wang X D, Wang S F, Zhao Q, Zhang M L, Qin J G, Chen L Q. 2018. Gut microbiota and its modulation for healthy farming of pacific white shrimp Litopenaeus vannamei. Reviews in Fisheries Science & Aquaculture, 26(3): 381-399.
DOI:10.1080/23308249.2018.1440530 |
Li T T, Long M, Ji C, Shen Z X, Gatesoupe F J, Zhang X J, Zhang Q Q, Zhang L L, Zhao Y L, Liu X H, Li A H. 2016. Alterations of the gut microbiome of largemouth bronze gudgeon (Coreius guichenoti) suffering from furunculosis. Scientific Reports, 6(1): 30606.
DOI:10.1038/srep30606 |
Li Y D, Zhou F L, Tang Y P, Huang J H, Yang L S, Jiang S, Yang Q B, Jiang S G. 2021. Variation in bacterial communities among stress-sensitive and stress-tolerant black tiger shrimp (Penaeus monodon) individuals. Aquaculture Research, 52(5): 2146-2159.
DOI:10.1111/are.15067 |
Liu Q, Long Y N, Li B, Zhao L L, Luo J, Xu L, Luo W, Du Z J, Zhou J, Yang S. 2020a. Rice-shrimp culture: a better intestinal microbiota, immune enzymatic activities, and muscle relish of crayfish (Procambarus clarkii) in Sichuan Province. Applied Microbiology and Biotechnology, 104(21): 9413-9420.
DOI:10.1007/s00253-020-10797-4 |
Liu X D, He X, An Z H, Sun W, Chen N, Gao X J, Li X X, Zhang X J. 2020b. Citrobacter freundii infection in red swamp crayfish (Procambarus clarkii) and host immune-related gene expression profiles. Aquaculture, 515: 734499.
DOI:10.1016/j.aquaculture.2019.734499 |
Magoč T, Salzberg S L. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27(21): 2957-2963.
DOI:10.1093/bioinformatics/btr507 |
Othman M, Agüero R, Lin H C. 2008. Alterations in intestinal microbial flora and human disease. Current Opinion in Gastroenterology, 24(1): 11-16.
DOI:10.1097/MOG.0b013e3282f2b0d7 |
Ott S J, Musfeldt M, Wenderoth D F, Hampe J, Brant O, Fölsch U R, Timmis K N, Schreiber S. 2004. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut, 53(5): 685-693.
DOI:10.1136/gut.2003.025403 |
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. 2016. VSEARCH: a versatile open source tool for metagenomics. PeerJ, 4: e2584.
DOI:10.7717/peerj.2584 |
Sekirov I, Russell S L, Antunes L C M, Finlay B B. 2010. Gut microbiota in health and disease. Physiological Reviews, 90(3): 859-904.
DOI:10.1152/physrev.00045.2009 |
Semova I, Carten J D, Stombaugh J, Mackey L C, Knight R, Farber S A, Rawls J F. 2012. Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host & Microbe, 12(3): 277-288.
DOI:10.1016/j.chom.2012.08.003 |
Serrano-Villar S, Rojo D, Martinez-Martinez, M, Deusch S, Vazquez-Castellanos J F, Bargiela R, Sainz T, Vera M, Moreno S, Estrada V, Jose Gosalbes M, Latorre A, Seifert J, Barbas C, Moya A, Ferrer M. 2016. Gut bacteria metabolism impacts immune recovery in HIV-infected individuals. Ebiomedicine, 8: 203-216.
DOI:10.1016/j.ebiom.2016.04.033 |
Shen G Q, Zhang X, Gong J, Wang Y, Huang P D, Shui Y, Xu Z H, Shen H S. 2020. Transcriptomic analysis of Procambarus clarkii affected by "Black May" disease. Scientific Reports, 10(1): 21225.
DOI:10.1038/s41598-020-78191-8 |
Shi H N, Walker A. 2004. Bacterial colonization and the development of intestinal defences. Canadian Journal of Gastroenterology and Hepatology, 18: 690421.
DOI:10.1155/2004/690421 |
Shui Y, Guan Z B, Liu G F, Fan L M. 2020. Gut microbiota of red swamp crayfish Procambarus clarkii in integrated crayfish-rice cultivation model. AMB Express, 10(1): 5.
DOI:10.1186/s13568-019-0944-9 |
Wang W, Wen B H, Gasparich G E, Zhu N N, Rong L W, Chen J X, Xu Z K. 2004. A spiroplasma associated with tremor disease in the Chinese mitten crab (Eriocheir sinensis). Microbiology, 150(9): 3035-3040.
DOI:10.1099/mic.0.26664-0 |
Watanabe M, Houten S M, Mataki C, Christoffolete M A, Kim B W, Sato H, Messaddeq N, Harney J W, Ezaki O, Kodama T, Schoonjans K, Bianco A C, Auwerx J. 2006. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature, 439(7075): 484-489.
DOI:10.1038/nature04330 |
Xiong J B, Wang K, Wu J F, Qiuqian L L, Yang K J, Qian Y X, Zhang D M. 2015. Changes in intestinal bacterial communities are closely associated with shrimp disease severity. Applied Microbiology and Biotechnology, 99(16): 6911-6919.
DOI:10.1007/s00253-015-6632-z |
Xu J, Mahowald M A, Ley R E, Lozupone C A, Hamady M, Martens E C, Henrissat B, Coutinho P M, Minx P, Latreille P, Cordum H, Van Brunt A, Kim K, Fulton R S, Fulton L A, Clifton S W, Wilson R K, Knight R D, Gordon J I. 2007. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biology, 5(7): e156.
DOI:10.1371/journal.pbio.0050156 |
Yue G H, Wang G L, Zhu B Q, Wang C M, Zhu Z Y, Lo L C. 2008. Discovery of four natural clones in a crayfish species Procambarus clarkii. International Journal of Biological Sciences, 4(5): 279-282.
|
Zeng T L, Ye Y F, Mu C K, Wang K, Li R H, Wang C L. 2016. Gut Microbiota and metabolic phenotype of Portunus trituberculatus. Chinese Journal of Analytical Chemistry, 44(12): 1867-1873.
DOI:10.1016/S1872-2040(16)60978-7 |
Zhang Y, Li Z Y, Kholodkevich S, Sharov A, Chen C, Feng Y J, Ren N Q, Sun K. 2020. Effects of cadmium on intestinal histology and microbiota in freshwater crayfish (Procambarus clarkii). Chemosphere, 242: 125105.
DOI:10.1016/j.chemosphere.2019.125105 |
Zhang Y, Sun K, Li Z Y, Chai X X, Fu X Y, Kholodkevich S, Kuznetsova T, Chen C, Ren N Q. 2021. Effescts of acute diclofenac exposure on intestinal histology, antioxidant defense, and microbiota in freshwater crayfish(Procambarus clarkii). Chemosphere, 263: 128130.
DOI:10.1016/j.chemosphere.2020.128130 |
Zhong Y D, Marungruang N, Fak F, Nyman M. 2015. Effects of two whole-grain barley varieties on caecal SCFA, gut microbiota and plasma inflammatory markers in rats consuming low- and high-fat diets. British Journal of Nutrition, 113(10): 1558-1570.
DOI:10.1017/S0007114515000793 |
Zhou Y T, Zhi F C. 2016. Lower level of Bacteroides in the gut microbiota is associated with inflammatory bowel disease: a meta-analysis. Biomed Research International, 2016: 5828959.
DOI:10.1155/2016/5828959 |