 2022, Vol. 40
2022, Vol. 40Institute of Oceanology, Chinese Academy of Sciences
Article Information
- LOU Fangrui, HAN Zhiqiang
- Full-length transcripts facilitates Portunus trituberculatus genome structure annotation
- Journal of Oceanology and Limnology, 40(5): 2042-2051
- http://dx.doi.org/10.1007/s00343-021-1248-x
Article History
- Received Jul. 27, 2021
- accepted in principle Oct. 28, 2021
- accepted for publication Nov. 28, 2021




2 Fishery College, Zhejiang Ocean University, Zhoushan 316022, China
The functional, physiological, and biosynthetic cellular states of crustacean are very complex, and the structural and functional genomics studies are fundamental for understanding the crustacean biology and essential to the access to high-quality genome resources. The high-throughput sequencing technologies stimulated the construction of reference genome resources for many crustaceans (Colbourne et al., 2011; Zhang et al., 2019). However, some reference genomes are often incomplete and insufficient to decode the annotation and structure (Choi et al., 2015; Shen-Gunther et al., 2016). Meanwhile, it seems that the encoding potential and gene expression regulation of crustaceans cannot be determined based on genomic information alone, because the post-transcriptional processing of precursor mRNAs is very diverse due to the presence of alternative splicing and polyadenylation (Kalsotra and Cooper, 2011; Elkon et al., 2013). The characteristics of transcriptome provides an opportunity to improve the accuracy and completeness of crustacean genome resources and elucidate the complexity of multiple biological mechanisms as the transcriptome information depicts the gene expression level and individual splice junction (Mortazavi et al., 2008; Wang et al., 2009).
Having obtained via high-throughput short-read sequencing (RNA-seq), transcriptome sequences of many crustaceans have been accumulated recent years (Xu et al., 2017; Lou et al., 2018). However, full-length reads beneficial for subsequent functional and transcriptional behavioral studies of crustacean important loci than short transcripts are recently considered as full-length transcripts can effectively predict the exon-intron structures, alternative splicing, alternative polyadenylation, and other genome structures (Ogihara et al., 2004; Soderlund et al., 2009). Since 2012, the third-generation sequencing platforms, such as Pacific Biosciences (PacBio) and Oxford Nanopore (ONT), have been gradually applied to long-read sequencing (Eid et al., 2009; Feng et al., 2015).
The swimming crab Portunus trituberculatus, one of the Asia's most valuable marine crustaceans, is an ideal model for elucidating crustacean genetic networks (Qi et al., 2013). Research into P. trituberculatus is facilitated by increasingly refined transcriptome knowledge of the morphological and physiological characteristics. The P. trituberculatus chromosome-level reference genome, assembled in 2020, used a sequencing strategy combining short-reads, ONT long-reads, and high-through chromosome conformation capture (Hi-C) sequencing data (Tang et al., 2020). Although the assembly effect of P. trituberculatus genome is relatively perfect, the defects in the ONT sequencing technology are apparent (Wyman et al., 2019). Meanwhile, short-reads used for P. trituberculatus genome annotation do not provide the full-length sequence of each RNA, limiting their utility for defining the genome annotation of P. trituberculatus.
We combined the Illumina and PacBio platforms to generate a more complete P. trituberculatus full-length transcriptome with rich data of full-length cDNA sequences that extends our knowledge of P. trituberculatus transcriptome. Our research confirmed the ability and reliability of long-reads in the discovery of full-length cDNA transcripts and novel genes/isoforms, which will improve genome annotation efficiency of P. trituberculatus.
2 MATERIAL AND METHOD 2.1 Animal specimenFemale P. trituberculatus specimen was collected from the coastal water of Zhoushan, China. P. trituberculatus was kept in aquarium for 4 days with fully aerated seawater. Next, P. trituberculatus was immediately anesthetized and tissues (gill, muscle, heart, and intestinal) were then rapidly sampled, snap-frozen in liquid nitrogen at -80 ℃.
2.2 Total RNA extractionThe total RNA of each tissue was extracted using the standard Trizol Reagent Kit (Huayueyang Biotech Co. Ltd., Beijing, China) following the manufacturer's protocol. Later, the RNAs from all tissues were pooled in equal amounts. The concentration and integrity of mix RNA were assessed using the NanoDrop 2000 system (Thermo Fisher Scientific, MA, USA) and the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). The mRNA was obtained by depleting rRNA of mix RNA using the RNA Purification Beads and then was cleaned three times using Beads Binding Buffer.
2.3 Illumina sequencing library construction and sequencingThe purified mRNA was incubated and used in turn to synthesize the first- and second-strand cDNA. Afterward, A-Tailing Control and Ligation Control were applied to the tailing and adapter ligation of the double stranded cDNA, respectively. All cDNA fragments were enriched to complete the libraries construction. After the libraries were diluted to 10 pmol/L, the Agilent 2100 Bioanalyzer was used for quantitative analysis, and the qualified libraries were sequenced on an Illumina HiSeq 2500 across one lane with paired-end 150 bp.
2.4 Single molecule real-time (SMRT) sequencing library construction and sequencingThe purified mRNA was incubated and then applied to synthesize the full-length cDNA required for sequencing using the SMARTerTM PCR cDNA Synthesis Kit (TaKaRa, USA), and then high-quality large-scale library amplification products were purified to 1–6 kb using the BluePippinTM Size-Selection System (Sage Science Inc., Beverly, MA, USA). The selected library was further undergone damage renovation, blunt end ligation, and addition of SMTBell adapters, which eventually form a SMRTbell template library. Finally, polymerase was added to the SMRTbell template and the resulted polymeride was fixed to the zero-mode waveguide (ZMW). One SMRT cell was prepared and used on the PacBio RSII platform for full-length transcriptome sequencing.
2.5 SMRT sequencing data processingThe SMRT analysis software v2.3 Suite (http://www.pacb.com/devnet/) was applied to filter out the low quality fragments in length < 50 bp and accuracy < 0.9 in the polymerase reads generated by the PacBio RSII platform according to the following parameters: readScore 0.90 and minLength 50. Subreads were obtained by interrupting the remaining high-quality polymerase reads at the adapter location and filtering out the adapter sequences. Subreads sequences < 50 bp were filtered using software SMRT v2.3 with parameters: minSubReadLength at 50, and the remaining subreads are identified as clean reads. The Iso-seq pipeline of software SMRTLink was used to extract the circular consensus (CCS) reads from the clean reads according to the following conditions: full passes > 1 and sequence accuracy > 0.90. CCS reads containing the correct 5' primers, 3' primers, and polyA tails were identified as full-length sequences, or otherwise were identified as non-full-length sequences. The insertion sequences of CCS reads were obtained by removing cDNA primer sequences and polyA sequences, and the direction of chain synthesis was determined according to the differences of primers at both ends of CCS sequences. These CCS reads were divided into full-length sequences, non-full-length sequences, chimeric sequences, and non-chimeric sequences. Next, the Iso-seq pipeline of software SMRTLink was applied to cluster those similar full-length non-chimeric sequences (multiple copies of the same transcript) to obtain consensus isoforms. The software Quiver was used to calibrate the consensus isoforms to obtain high-quality (accuracy > 99%) and low-quality transcripts (Li et al., 2018). To improve low-quality transcripts accuracy, low-quality consensus sequences were corrected using clean RNA-seq data based on software Proovread (Hackl et al., 2014). Strict parameters were set in the clustering process of full-length transcripts. In order to obtain high quality consensus sequences, the possibility of multi-copy sequences of the same transcript being divided into different clusters is higher than the possibility of randomly clustering two copy sequences that do not belong to the same transcript, which inevitably leads to redundant sequences. Meanwhile, the 5' end of some reads will be degraded in the full-length transcriptome sequencing process, resulting in different copies of the same transcript cannot be clustered together. Software CD-HIT (Li and Godzik, 2006) can combine sequences with high similarity, thus it was used to remove redundant sequences from high-quality transcripts (Fig. 1).

|
| Fig.1 Remove redundancy process of consensus sequences The green oval represents the same transcript with differences at the 5' end. |
Due to relatively low conservativeness of repetitive sequences among species, it is necessary to construct specific repeat database when predicting repetitive sequences for P. trituberculatus. In this study, software LTR_FINDER (Xu and Wang, 2007) and RepeatScout (Price et al., 2005) were applied to construct the P. trituberculatus transcriptome repeating sequence database based on the principle of structure prediction and de novo prediction. The database classified by software PASTEClassifier (Hoede et al., 2014) was merged with Repbase database (Jurka et al., 2005) to form final repeating sequence database. Finally, software RepeatMasker (Tarailo-Graovac and Chen, 2009) was used to predict the repetitive sequence based on the constructed repetitive sequence database.
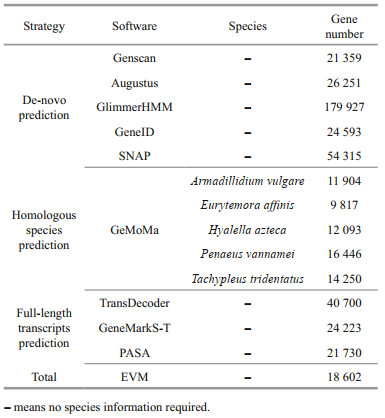
2.6.2 Gene structure predictionThree strategies, i.e., de novo prediction, homologous species prediction, and full-length transcripts prediction, were applied to predict the gene structure. In this study, de novo prediction was performed using software Genscan (Burge and Karlin, 1997), Augustus (Stanke and Waack, 2003), GlimmerHMM (Majoros et al., 2004), GeneID (Blanco et al., 2007), and SNAP (Korf, 2004). GeMoMa software (Keilwagen et al., 2016) was used to carry out homologous species prediction based on Armadillidium vulgare, Eurytemora affinis, Hyalella Azteca, Penaeus vannamei, and Tachypleus tridentatus. Additionally, software TransDecoder (http://transdecoder.github.io), GeneMarkS-T (Tang et al., 2015), and PASA (Campbell et al., 2006) were used to full-length transcripts prediction. Finally, software EVM (Haas et al., 2008) was used to integrate the predicted results from three strategies.
2.6.3 Non-coding RNAs predictionNon-coding RNAs are those who do not encode proteins, including miRNA, rRNA, tRNA, and other RNAs with known functions. According to the structural characteristics of different non-coding RNAs, different strategies were adopted to predict different non-coding RNAs. In this study, we have identified the microRNA and rRNA based on the Rfam database using Blastn (Griffiths-Jones et al., 2005). Meanwhile, software tRNAscan-SE (Lowe and Eddy, 1997) was used to predict the tRNA.
2.6.4 Pseudogenes annotationThe sequence of pseudogenes is similar to those of functional genes, but pseudogene loses its original function due to insertion, deletion, and other mutations. We used the predicted protein sequences and searched for homologous gene sequences (possible genes) on the P. trituberculatus genome by BLAT (Kent, 2002) alignment. Furthermore, pseudogenes were obtained by using software Genewise (Birney et al., 2004) to search for immature termination codon and frame shift mutation in gene sequence.
2.6.5 Gene function annotationAll the predicted gene sequences were applied to analyze the gene ontology and orthologous classifications based on NR, KOG, GO, KEGG, and TrEMBL database using the BLAST (Altschul et al., 1990) and the parameter E-value < 0.00001. Meanwhile, software InterProScan (Zdobnov and Apweiler, 2001) was applied to annotate the sequence motif based on the PROSITE, HAMAP, Pfam, PRINTS, ProDom, SMART, TIGRFAMs, PIRSF, SUPERFAMILY, CATH-Gene3D, and PANTHER databases.
3 RESULT 3.1 P. trituberculatus full-length transcriptome sequencing using PacBio platformHigh-quality total RNA was extracted from the multiple tissues of P. trituberculatus and full-length transcriptome sequencing was then performed on the PacBio RSII platform. After filtering out the low quality sequencing data and removing the adapter sequence, 42.6-Gb subreads were obtained form 1 SMRT cells. After filtering out subreads below 50 bp in length, 23-Gb clean subreads were obtained in this study. Furthermore, 366 770 CCS reads were screened out from the clean subreads, corresponding to 847 034 414 bp. Meanwhile, the mean read length and the mean sequencing depth of CCS reads was 2 309 bp and 39 passes, respectively, among which 261 362 (71.26%) were considered as full-length non-chimeric sequences. Similar sequences in all the full-length non-chimeric sequences were clustered into 15 832 consistent isoforms, with the mean length of 2 493 bp. After correcting the consistent isoforms, 15 710 (99.23%) high-quality and 109 low-quality transcripts were obtained. The length distribution of CCS, full-length non-chimeric reads, and consensus isoforms are shown in Fig. 2. Finally, CD-HIT (Li and Godzik, 2006) was applied to remove redundant sequences from high-quality transcripts, and 9 694 non-redundant full-length transcripts were yielded and used for the following analyses.

|
| Fig.2 The length distribution of CCS (a), full-length non-chimeric reads (b), and consensus isoforms (c) |
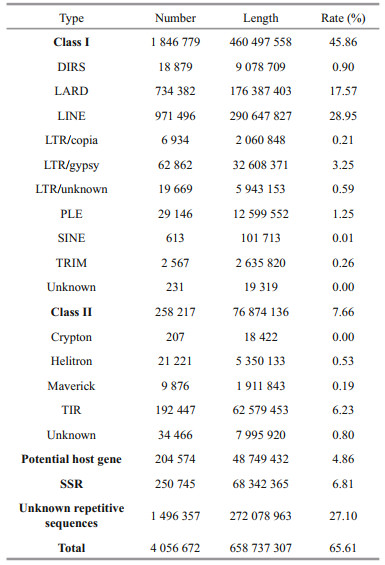
The cumulative sequences of repetitive sequences identified in the P. trituberculatus full-length transcriptome occupied 65.61% of the genome, corresponding to 658 737 307 bp (Table 1). Among all the repetitive sequences, simple sequence repeats (SSRs) and potential host gene occupied 6.81% and 4.86% of the P. trituberculatus genome in total length of 68 342 365 bp and 48 749 432 bp, respectively. Meanwhile, the transposable elements (TEs) occupied 53.52% of the P. trituberculatus genome in total length of 537 371 694 bp. The detected TEs included mainly retrotransposons (Class Ⅰ; 460 497 558 bp) and DNA transposons (Class Ⅱ; 76 874 136 bp). Class Ⅰ retrotransposons could be divided into eight groups, including DIRS (9 078 709 bp), LARD (176 387 403 bp), LINE (290 647 827 bp), LTR (40 612 372 bp), PLE (12 599 552 bp), SINE (101 713 bp), TRIM (2 635 820 bp), and 19 319-bp unknown retrotransposons. Class Ⅱ DNA transposons can be divided into five groups, including Crypton (18 422 bp), Helitron (5 350 133 bp), Maverick (1 911 843 bp), TIR (62 579 453 bp), and 7 995 920-bp unknown DNA transposons. Additionally, 272 078 963-bp repetitive sequences could not be classified at the present study.
In the present study, three strategies were applied to predict the coding gene. Results showed that the number of genes predicted by different analysis strategies was different (Table 2) and a total of 17 519, 14 633, and 12 110 genes were predicted based on de novo prediction, homologous species prediction, and full-length transcripts prediction (Fig. 3). In order to improve the accuracy of gene prediction, all the gene prediction results were integrated and a total of 18 602 genes were obtained in total length of 219 006 379 bp and mean length of 11 773.27 bp, respectively. Meanwhile, we also predicted 123 333 exons, 104 731 introns, and 119 274 coding genes across all genes, with a total length of 33 951 173 bp, 185 055 206 bp, and 26 236 140 bp, respectively.

|
| Fig.3 The integrated gene prediction results based on three strategies |
In this study, 28 686 non-coding RNAs were predicted in the full-length transcriptome of P. trituberculatus. Among all the non-coding RNAs, there are 36 miRNAs, 216 rRNAs, and 28 434 tRNAs were predicted, belonging to 23 miRNA families, 4 rRNA families, and 25 tRNA families, respectively.
3.3.3 PseudogenesAfter looking for homologous gene sequences on the P. trituberculatus genome and then identifying the immature termination codon and frameshift mutations, 1 407 pseudogenes were predicted in the P. trituberculatus full-length transcriptome in total length of 3 368 546 bp on average of 2 394.13 bp.
3.3.4 Gene function annotationTo obtain the gene functional information, all the predicted gene sequences were applied to compare with the sequences of five databases. Results show that 78.24% (14 554/18 602) genes were annotated in five databases. Of all homology searches, 14 460 (77.73%), 10 211 (54.89%), 5 412 (29.09%), 7 314 (39.32%), and 14 448 (77.67%) genes had significant matches with sequences in the NR, KOG, GO, KEGG, and TrEMBL database, respectively. After annotation, 740 motif and 26 434 domain were also be found in the gene sequences.
4 DISCUSSION 4.1 Improvement of annotated information of P. trituberculatus genomeTang et al. (2020) constructed a P. trituberculatus chromosomal-level genome and the genome assembly quality has been rigorously validated in a variety of methods. However, short transcripts used in genome annotation are difficult to provide full-length transcript isoforms for each RNA, limiting their utility for defining alternative splicing and alternative polyadenylation and perfecting genomic information (Au et al., 2013). Meanwhile, the quantification of expression levels are complicated by multiple amplification steps in the library preparation of RNA-seq (Sharon et al., 2013). On the other hand, short-reads transcribed by highly repetitive regions or very similar members of multigene families can also be misassembled (Schliesky et al., 2012; Li et al., 2014). Considering the shortcomings of short transcripts in the application of P. trituberculatus genome annotation, the well-characterized full-length transcript tags can compensate for the above deficiencies, and ultimately are beneficial for behavioral studies on subsequent function and transcription of important loci of P. trituberculatus. Because of the longer reads, both PacBio (the read length is up to 60 kb) and ONT (the read length is up to 1 Mb) platforms can capture complete transcripts from end to end (Wyman et al., 2019). In fact, PacBio platform yields has increased dramatically in recent years, with generating up to 8 million reads per SMRT cell on the Sequel 2, far more than original RSII machines (0.15 million reads). Similar yield increases have been reported for ONT platform (Wyman et al., 2019). Meanwhile, both two platforms have the advantage of representing single molecules rather than amplified clusters, thus facilitating isoforms sequencing (Eid et al., 2009). However, it is worth noting that long-reads sequencing from the third-generation sequencing platforms also has some disadvantages, such as high indel and mismatch error rates, especially the ONT platform (Eid et al., 2009). Compared with the ONT platform, the random errors in the sequencing process of the PacBio platform can be alleviated by self-correction of long reads, correction of short reads and some computational methods (Rhoads and Au, 2015; Lou et al., 2020). Unfortunately, most of the available tools for analyzing long-reads were not specifically designed for direct quantification of long-reads. Considering that the ONT platform has long reads, and the PacBio platform has high reads accuracy, we speculate that using PacBio platforms can achieve more efficient P. trituberculatus full-length transcripts and aim to improve the annotated information of P. trituberculatus genome.
4.2 High proportion of repetitive sequencesA total of 658.74 Mb of repetitive sequences were identified and accounting for 65.61% of the P. trituberculatus genome, which is greater than 54.52% reported by Tang et al. (2020). Therefoore, we hypothesized that different transcripts of the same gene may increase the number of repetitive sequences. Previous association analysis by Gao et al. (2018) between the number of whole-genome repetitive sequences and genome size in 44 plants and 68 vertebrates suggested that the proportion of repetitive sequences was positively correlated with genome size. Therefore, the high proportion (either 65.61% or 54.52%) of repetitive sequences also confirmed the large genome size (the genome assembly size was 1.00 Gb by Tang et al., 2020) and high complexity of P. trituberculatus. Meanwhile, we also found the TEs occupied 53.52% of the P. trituberculatus genome. Previous studies have speculated that an increase in the number and type of TEs would contribute to the genome size expansion and evolution (Cordaux and Batzer, 2009; Sun et al., 2012). Additionally, TEs have been found to play a critical role in some biological events, such as organism development (Kano et al., 2009; Garcia-Perez et al., 2016), regulation (Elbarbary et al., 2016), differentiation (Morales-Hernández et al., 2016), and as promoters to activate the transcription process (Faulkner et al., 2009; Mita and Boeke, 2016). Therefore, we hypothesize that such abundant TE may help regulate some biological behavior of P. trituberculatus.
4.3 Optimization of P. trituberculatus genetic structure informationIt is worth noting that the genome structure prediction is often inaccurate due to the limitations of the short transcripts, so we used full-length transcripts to predict genome structure more accurately. In this study, three prediction strategies were used to annotate protein-coding genes and ensure the accuracy of the prediction results. We merged the prediction results and obtained 18 602 protein-coding genes, which is greater than previously predicted number (16 791; Tang et al., 2020) based on genomic data at the chromosomal level and indicate more undiscovered genes. In fact, full-length transcripts have been shown helpful in the prediction of novel genes and novel isoforms of Tachypleus tridentatus (Lou et al., 2020). We believe that full-length transcripts will be necessary for future studies of novel genes and isoforms, although the genome annotated file in generic feature format (GFF) format has not been released to limit the prediction of novel genes and novel isoforms of P. trituberculatus.
Meanwhile, we predicted 28 686 non-coding RNAs and 1 407 pseudogenes in the P. trituberculatus full-length transcripts. Many studies have concluded that non-coding RNAs and pseudogenes also play important roles in the regulation of biological growth and development, abiotic stress, and other physiological functions (Schiff et al., 1985; Liu et al., 2013; Chen and Ge, 2017). Additionally, non-coding RNAs and pseudogenes appear to be more species-specific and may ultimately provide more appropriate evidence for the study of biological evolution. Therefore, we have every reason to believe that the non-coding RNAs and pseudogenes predicted in this study will contribute to the further biological research of P. trituberculatus.
5 CONCLUSIONIn this study, we have assembled the P. trituberculatus full-length transcriptome based on SMRT sequencing technology. A total of 9 694 full-length transcripts were generated successfully after filtering out low-quality sequencing reads, self-correcting, and de-redundancy. We also successfully predicted the repetitive sequences, coding genes, non-coding RNAs, and pseudogenes. These results not only help to refine the P. trituberculatus genomic annotation information, but also provide basic resources for the future evolution and physiological regulation researches of P. trituberculatus.
6 DATA AVAILABILITY STATEMENTAll subreads in bam format were released in the NCBI Sequence Read Archive under BioProject number PRJNA749655, with accession number of SRR15248879.
Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. 1990. Basic local alignment search tool. Journal of Molecular Biology, 215(3): 403-410.
DOI:10.1016/S0022-2836(05)80360-2 |
Au K F, Sebastiano V, Afshar P T, Durruthy J D, Lee L, Williams B A, van Bakel H, Schadt E E, Reijo-Pera R A, Underwood J G, Wong W H. 2013. Characterization of the human ESC transcriptome by hybrid sequencing. Proceedings of the National Academy of Sciences of the United States of America, 110(50): E4821-E4830.
DOI:10.1073/pnas.1320101110 |
Birney E, Clamp M, Durbin R. 2004. GeneWise and genomewise. Genome Research, 14(5): 988-995.
DOI:10.1101/gr.1865504 |
Blanco E, Parra G, Guigó R. 2007. Using geneid to identify genes. Current Protocols in Bioinformatics, Chapter 4: Unit 4.3, https://doi.org/10.1002/0471250953.bi0403s18.
|
Burge C, Karlin S. 1997. Prediction of complete gene structures in human genomic DNA. Journal of Molecular Biology, 268(1): 78-94.
DOI:10.1006/jmbi.1997.0951 |
Campbell M A, Haas B J, Hamilton J P, Mount S M, Buell C R. 2006. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics, 7: 327.
DOI:10.1186/1471-2164-7-327 |
Chen L, Ge S. 2017. A brief introduction of noncoding RNA research. Chinese Science Bulletin, 62(27): 3236-3244.
(in Chinese with English abstract) DOI:10.1360/N972017-00384 |
Choi J W, Chung W H, Lee K T, Cho E S, Lee S W, Choi B H, Lee S H, Lim W, Lim D, Lee Y G, Hong J K, Kim D W, Jeon H J, Kim J, Kim N, Kim T H. 2015. Whole-genome resequencing analyses of five pig breeds, including Korean wild and native, and three European origin breeds. DNA Research, 22(4): 259-267.
DOI:10.1093/dnares/dsv011 |
Colbourne J K, Pfrender M E, Gilbert D, Thomas W K, Tucker A, Oakley T H, Tokishita S, Aerts A, Arnold G J, Basu M K, Bauer D J, Cáceres C E, Carmel L, Casola C, Choi J H, Detter J C, Dong Q F, Dusheyko S, Eads B D, Fröhlich T, Geiler-Samerotte K A, Gerlach D, Hatcher P, Jogdeo S, Krijgsveld J, Kriventseva E V, Kültz D, Laforsch C, Lindquist E, Lopez J, Manak J R, Muller J, Pangilinan J, Patwardhan R P, Pitluck S, Pritham E J, Rechtsteiner A, Rho M, Rogozin I B, Sakarya O, Salamov A, Schaack S, Shapiro H, Shiga Y, Skalitzky C, Smith Z, Souvorov A, Sung W, Tang Z J, Tsuchiya D, Tu H, Vos H, Wang M, Wolf Y I, Yamagata H, Yamada T, Ye Y Z, Shaw J R, Andrews J, Crease T J, Tang H X, Lucas S M, Robertson H M, Bork P, Koonin E V, Zdobnov E M, Grigoriev I V, Lynch M, Boore J L. 2011. The ecoresponsive genome of Daphnia pulex. Science, 331(6017): 555-561.
DOI:10.1126/science.1197761 |
Cordaux R, Batzer M A. 2009. The impact of retrotransposons on human genome evolution. Nature Reviews Genetics, 10(10): 691-703.
DOI:10.1038/nrg2640 |
Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G N, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S. 2009. Real-time DNA sequencing from single polymerase molecules. Science, 323(5910): 133-138.
DOI:10.1126/science.1162986 |
Elbarbary R A, Lucas B A, Maquat L E. 2016. Retrotransposons as regulators of gene expression. Science, 351(6274): aac7247.
DOI:10.1126/science.aac7247 |
Elkon R, Ugalde A P, Agami R. 2013. Alternative cleavage and polyadenylation: extent, regulation and function. Nature Reviews Genetics, 14(7): 496-506.
DOI:10.1038/nrg3482 |
Faulkner G J, Kimura Y, Daub C O, Wani S, Plessy C, Irvine K M, Schroder K, Cloonan N, Steptoe A L, Lassmann T, Waki K, Hornig N, Arakawa T, Takahashi H, Kawai J, Forrest A R R, Suzuki H, Hayashizaki Y, Hume D A, Orlando V, Grimmond S M, Carninci P. 2009. The regulated retrotransposon transcriptome of mammalian cells. Nature Genetics, 41(5): 563-571.
DOI:10.1038/ng.368 |
Feng Y X, Zhang Y C, Ying C F, Wang D Q, Du C L. 2015. Nanopore-based fourth-generation DNA sequencing technology. Genomics, Proteomics & Bioinformatics, 13(1): 4-16.
DOI:10.1016/j.gpb.2015.01.009 |
Gao S H, Yu H Y, Wu S Y, Wang S, Geng J N, Luo Y F, Hu S N. 2018. Advances of sequencing and assembling technologies for complex genomes. Hereditas, 40(11): 944-963.
(in Chinese with English abstract) DOI:10.16288/j.yczz.18-255 |
Garcia-Perez J L, Widmann T J, Adams I R. 2016. The impact of transposable elements on mammalian development. Development, 143(22): 4101-4114.
DOI:10.1242/dev.132639 |
Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy S R, Bateman A. 2005. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Research, 33(suppl_1): D121-D124.
DOI:10.1093/nar/gki081 |
Haas B J, Salzberg S L, Zhu W, Pertea M, Allen J E, Orvis J, White O, Buell C R, Wortman J R. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biology, 9(1): R7.
DOI:10.1186/gb-2008-9-1-r7 |
Hackl T, Hedrich R, Schultz J, Förster F. 2014. proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics, 30(21): 3004-3011.
DOI:10.1093/bioinformatics/btu392 |
Hoede C, Arnoux S, Moisset M, Chaumier T, Inizan O, Jamilloux V, Quesneville H. 2014. PASTEC: an automatic transposable element classification tool. PLoS One, 9(5): e91929.
DOI:10.1371/journal.pone.0091929 |
Jurka J, Kapitonov V V, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. 2005. Repbase update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research, 110(1-4): 462-467.
DOI:10.1159/000084979 |
Kalsotra A, Cooper T A. 2011. Functional consequences of developmentally regulated alternative splicing. Nature Reviews Genetics, 12(10): 715-729.
DOI:10.1038/nrg3052 |
Kano H, Godoy I, Courtney C, Vetter M R, Gerton G L, Ostertag E M, Kazazian H H Jr. 2009. L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes & Development, 23(11): 1303-1312.
DOI:10.1101/gad.1803909 |
Keilwagen J, Wenk M, Erickson J L, Schattat M H, Grau J, Hartung F. 2016. Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9): e89.
DOI:10.1093/nar/gkw092 |
Kent W J. 2002. BLAT—the BLAST-like alignment tool. Genome Research, 12(4): 656-664.
DOI:10.1101/gr.229202 |
Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics, 5: 59.
DOI:10.1186/1471-2105-5-59 |
Li B, Fillmore N, Bai Y S, Collins M, Thomson J A, Stewart R, Dewey C N. 2014. Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biology, 15(12): 553.
DOI:10.1186/s13059-014-0553-5 |
Li W Z, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics, 22(13): 1658-1659.
DOI:10.1093/bioinformatics/btl158 |
Li Y, Fang C C, Fu Y H, Hu A, Li C C, Zou C, Li X Y, Zhao S H, Zhang C J, Li C C. 2018. A survey of transcriptome complexity in Sus scrofa using single-molecule long-read sequencing. DNA Research, 25(4): 421-437.
DOI:10.1093/dnares/dsy014 |
Liu H, Zou C, Lin F. 2013. Identification and function analysis of pseudogenes. Chinese Journal of Biotechnology, 29(5): 551-567.
(in Chinese with English abstract) DOI:10.13345/j.cjb.2013.05.013 |
Lou F R, Song N, Han Z Q, Gao T X. 2020. Single-molecule real-time (SMRT) sequencing facilitates Tachypleus tridentatus genome annotation. International Journal of Biological Macromolecules, 147: 89-97.
DOI:10.1016/j.ijbiomac.2020.01.029 |
Lou F R, Yang T Y, Han Z Q, Gao T X. 2018. Transcriptome analysis for identification of candidate genes related to sex determination and growth in Charybdis japonica. Gene, 677: 10-16.
DOI:10.1016/j.gene.2018.07.044 |
Lowe T M, Eddy S R. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research, 25(5): 955-964.
DOI:10.1093/nar/25.5.955 |
Majoros W H, Pertea M, Salzberg S L. 2004. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics, 20(16): 2878-2879.
DOI:10.1093/bioinformatics/bth315 |
Mita P, Boeke J D. 2016. How retrotransposons shape genome regulation. Current Opinion in Genetics & Development, 37: 90-100.
DOI:10.1016/j.gde.2016.01.001 |
Morales-Hernández A, González-Rico F J, Román A C, Rico-Leo E, Alvarez-Barrientos A, Sánchez L, Macia á, Heras S R, García-Pérez J L, Merino J M, Fernández-Salguero P M. 2016. Alu retrotransposons promote differentiation of human carcinoma cells through the aryl hydrocarbon receptor. Nucleic Acids Research, 44(10): 4665-4683.
DOI:10.1093/nar/gkw095 |
Mortazavi A, Williams B A, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods, 5(7): 621-628.
DOI:10.1038/nmeth.1226 |
Ogihara Y, Mochida K, Kawaura K, Murai K, Seki M, Kamiya A, Shinozaki K, Carninci P, Hayashizaki Y, Shin-I T, Kohara Y, Yamazaki Y. 2004. Construction of a full-length cDNA library from young spikelets of hexaploid wheat and its characterization by large-scale sequencing of expressed sequence tags. Genes & Genetic Systems, 79(4): 227-232.
DOI:10.1266/ggs.79.227 |
Price A L, Jones N C, Pevzner P A. 2005. De novo identification of repeat families in large genomes. Bioinformatics, 21(suppl_1): i351-i358.
DOI:10.1093/bioinformatics/bti1018 |
Qi J B, Gu X L, Ma L B, Qiao Z G, Chen K. 2013. The research progress on food organism culture and technology utilization in crab seed production in ponds in China. Agricultural Sciences, 4(10): 563-569.
DOI:10.4236/as.2013.410076 |
Rhoads A, Au K F. 2015. PacBio sequencing and its applications. Genomics, Proteomics & Bioinformatics, 13(5): 278-289.
DOI:10.1016/j.gpb.2015.08.002 |
Schiff C, Milili M, Fougereau M. 1985. Functional and pseudogenes are similarly organized and may equally contribute to the extensive antibody diversity of the IgVHⅡ family. The EMBO Journal, 4(5): 1225-1230.
DOI:10.1002/j.1460-2075.1985.tb03764.x |
Schliesky S, Gowik U, Weber A P M, Bräutigam A. 2012. RNA-Seq assembly-are we there yet?. Frontiers in Plant Science, 3: 220.
DOI:10.3389/fpls.2012.00220 |
Sharon D, Tilgner H, Grubert F, Snyder M. 2013. A single-molecule long-read survey of the human transcriptome. Nature Biotechnology, 31(11): 1009-1014.
DOI:10.1038/nbt.2705 |
Shen-Gunther J, Wang C M, Poage G M, Lin C L, Perez L, Banks N A, Huang T H M. 2016. Molecular Pap smear: HPV genotype and DNA methylation of ADCY8, CDH8, and ZNF582 as an integrated biomarker for high-grade cervical cytology. Clinical Epigenetics, 8(1): 96.
DOI:10.1186/s13148-016-0263-9 |
Soderlund C, Descour A, Kudrna D, Bomhoff M, Boyd L, Currie J, Angelova A, Collura K, Wissotski M, Ashley E, Morrow D, Fernandes J, Walbot V, Yu Y. 2009. Sequencing, mapping, and analysis of 27, 455 maize full-length cDNAs. PloS Genetics, 5(11): e1000740.
DOI:10.1371/journal.pgen.1000740 |
Stanke M, Waack S. 2003. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics, 19(suppl_2): ii215-ii225.
DOI:10.1093/bioinformatics/btg1080 |
Sun C, Shepard D B, Chong R A, Arriaza J L, Hall K, Castoe T A, Feschotte C, Pollock D D, Mueller R L. 2012. LTR retrotransposons contribute to genomic gigantism in Plethodontid salamanders. Genome Biology and Evolution, 4(2): 168-183.
DOI:10.1093/gbe/evr139 |
Tang B P, Zhang D Z, Li H R, Jiang S H, Zhang H B, Xuan F J, Ge B M, Wang Z F, Liu Y, Sha Z L, Cheng Y X, Jiang W, Jiang H, Wang Z K, Wang K, Li C F, Sun Y, She S S, Qiu Q, Wang W, Li X Z, Li Y X, Liu Q N, Ren Y D. 2020. Chromosome-level genome assembly reveals the unique genome evolution of the swimming crab (Portunus trituberculatus). GigaScience, 9(1): giz161.
DOI:10.1093/gigascience/giz161 |
Tang S Y Y, Lomsadze A, Borodovsky M. 2015. Identification of protein coding regions in RNA transcripts. Nucleic Acids Research, 43(12): e78.
DOI:10.1093/nar/gkv227 |
Tarailo-Graovac M, Chen N S. 2009. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics, Chapter 4: Unit 4.10, https://doi.org/10.1002/0471250953.bi0410s25.
|
Wang Z, Gerstein M, Snyder M. 2009. RNA-seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics, 10(1): 57-63.
DOI:10.1038/nrg2484 |
Wyman D, Balderrama-Gutierrez G, Reese F, Jiang S, Rahmanian S, Forner S, Matheos D, Zeng W H, Williams B, Trout D, England W, Chu S H, Spitale R C, Tenner A J, Wold B J, Mortazavi A. 2019. A technology-agnostic long-read analysis pipeline for transcriptome discovery and quantification. BioRxiv: 672931.
DOI:10.1101/672931 |
Xu Y, Li X G, Deng Y F, Lu Q P, Yang Y J, Pan J L, Ge J C, Xu Z Q. 2017. Comparative transcriptome sequencing of the hepatopancreas reveals differentially expressed genes in the precocious juvenile Chinese mitten crab, Eriocheir sinensis (Crustacea: Decapoda). Aquaculture Research, 48(7): 3645-3656.
DOI:10.1111/are.13189 |
Xu Z, Wang H. 2007. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research, 35(suppl_2): W265-W268.
DOI:10.1093/nar/gkm286 |
Zdobnov E M, Apweiler R. 2001. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics, 17(9): 847-848.
DOI:10.1093/bioinformatics/17.9.847 |
Zhang X J, Yuan J B, Sun Y M, Li S H, Gao Y, Yu Y, Liu C Z, Wang Q C, Lv X J, Zhang X X, Ma K Y, Wang X B, Lin W, Wang L, Zhu X L, Zhang C S, Zhang J S, Jin S J, Yu K J, Kong J, Xu P, Chen J, Zhang H B, Sorgeloos P, Sagi A, Alcivar-Warren A, Liu Z J, Wang L, Ruan J, Chu K H, Liu B, Li F H, Xiang J H. 2019. Penaeid shrimp genome provides insights into benthic adaptation and frequent molting. Nature Communications, 10(1): 356.
DOI:10.1038/s41467-018-08197-4 |