 2022, Vol. 40
2022, Vol. 40Institute of Oceanology, Chinese Academy of Sciences
Article Information
- LI Mengyu, LI Yuqiang, XING Tengfei, LI Yulong, LIU Jinxian
- Microsatellite marker development and population genetic analysis revealed high connectivity between populations of a periwinkle Littoraria sinensis (Philippi, 1847)
- Journal of Oceanology and Limnology, 40(3): 1097-1109
- http://dx.doi.org/10.1007/s00343-021-1079-9
Article History
- Received Mar. 9, 2021
- accepted in principle May. 9, 2021
- accepted for publication Jun. 20, 2021




2 Laboratory for Marine Ecology and Environmental Science, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao 266237, China;
3 Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao 266071, China;
4 University of Chinese Academy of Sciences, Beijing 100049, China
The rocky intertidal zone, one of the most intensively studied ecosystems globally, is subject to environmental challenges posed by both aquatic and aerial climatic regimes (Raffaelli and Paine, 1995). It is an important coastal system, which is among the most artificially and heavily disturbed marine ecosystems (Menge, 2000). Periwinkles are important players in shaping the structure of intertidal communities (Apolinário et al., 1999). They can prey on other mobile predators and compete for space with algae and mussels on the hard-bottom intertidal zone (Cubit, 1984). The littorinid genus Littoraria is closely associated with rocky shores and mangroves (Reid et al., 2010).Littoraria sinensis(Gastropoda: Littorinidae), a periwinkle species, is one of the most conspicuous and abundant gastropods in the northwestern Pacific and distributed on the coast of Korea, Japan, and China. L. sinensis is a newly described species from Littoraria articulata based on a morphology analysis (Reid, 2001). As an intertidal dominant species, L. sinensis could play a significant ecological role in the intertidal communities (Reid, 1989).
Littoraria sinensis inhabits hard bottom habitat including rocky shores, trunks at the seaward edge of mangrove forests and artificial structures in the intertidal zone (Dong et al., 2016). The estimated planktonic larval duration of L. sinensis was 3–10 weeks (Reid, 2001). In the distribution range of L. sinensis, the increasing of artificial structures along the coasts of Jiangsu Province, China, provided new suitable habitats for L. sinensis and other intertidal marine invertebrates; and changed the intertidal communities (Dong et al., 2016). Before the construction of the artificial structures, the large mudflat of this area was probably an effective barrier to larval dispersal. For most benthic marine species, dispersal and exchange of individuals among populations occur primarily during the pelagic larval stage (Cowen and Sponaugle, 2009). Defining dispersal patterns of marine organisms is challenging since it could be affected by many factors including survival and development rates of larvae (Doropoulos et al., 2016), hydrodynamic regimes, and seascapes (Cowen et al., 2007; Cowen and Sponaugle, 2009). These challenges could be overcome by using molecular markers to evaluate the population dynamics of marine benthic species (Selkoe et al., 2016). Phylogenetic studies including L. sinensis have been conducted using molecular sequences, such as nuclear (28S rRNA) and mitochondrial (12S rRNA, COI) (Reid et al., 2010). In addition, a study using mitochondrial COI sequences demonstrated significant genetic differentiation between north and south populations of L. sinensis (Dong et al., 2016). The newly formed populations on the artificial structures south of Wanggangzha (33°10′N, 120°50′E) were significantly different from north natural populations and shared high genetic similarity with populations south of the Changjiang (Yangtze) River estuary (Dong et al., 2016). In addition, the artificial structures have promoted the northward distribution range shift of L. sinensis across the Changjiang River estuary. However, the power of the molecular markers adopted in Dong et al. (2016) for tracing the source of individuals in the newly formed populations is limited due to the low intraspecific variability and maternal inheritance of mitochondrial COI.
Higher-resolution molecular markers, such as microsatellites/simple sequence repeat (SSRs), may help to better understand the population patterns and draw convincing conclusions of phylogeographical structure (Selkoe and Toonen, 2006). Microsatellites are widely distributed throughout the genome, and are co-dominant and highly polymorphic (Estoup et al., 1993). It is one of the most widely used markers in molecular genetic applications, including population genetic analysis, germplasm conservation, forensic genetics, parentage analysis and the study of genealogy (Sharma et al., 2007; Schoebel et al., 2013; Flanagan and Jones, 2019). Microsatellite development was promoted by the high throughput and low-cost Next-Generation Sequencing (NGS) technologies, especially in non-model organisms with limited genomic information (Schoebel et al., 2013; Lopez-Marquez et al., 2018; Rosa et al., 2019). Microsatellites have been developed for several Littorinidae species (Tie et al., 2000; Kennington et al., 2008; Carvalho et al., 2015). However, most of them adopted the traditional methods which are laborious, time-consuming, and expensive. For the genus Littoraria, no microsatellite markers have been developed and the genomic resources of this genus are also lacking.
We investigated the population genetic structure of L. sinensis using newly developed microsatellite markers developed by the whole-genome shotgun sequencing approach, and conducted population genetic analyses of 15 geographic populations of L. sinensis along the coast of China using 12 polymorphic microsatellites, to clarify the population genetic structure across the distribution of L. sinensis, to check the source of the newly founded populations along the coast of Jiangsu Province, to improve our understanding of the larval dispersal process of marine intertidal organisms, and to provide references to the ecological connectivity of this region.
2 MATERIAL AND METHOD 2.1 Sampling and DNA extractionA total of 351 individuals of L. sinensis were collected from 15 geographic populations from natural rocky intertidal habitats and artificial structures along the coast of China (Table 1). Foot-muscle tissue was dissected and preserved in 95% ethanol. Genomic DNA was isolated using standard phenol-chloroform extraction protocol and DNA quality was checked using 1% agarose electrophoresis and Nanodrop 2000c spectrophotometer.
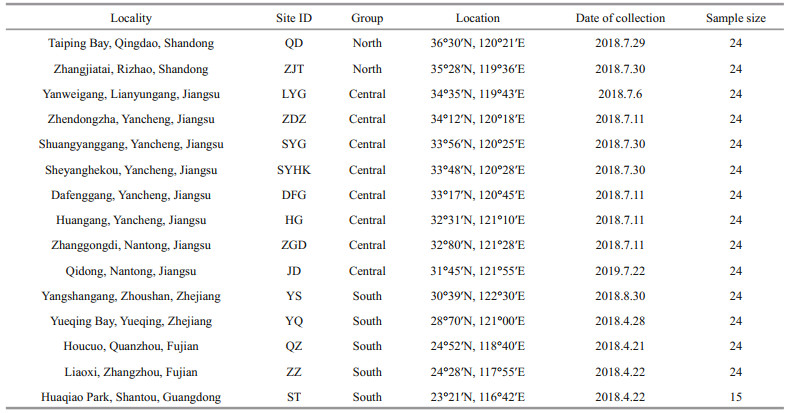
Foot-muscle tissue of one individual from Zhangjiatai (Rizhao, Shandong Province) was selected for whole-genome shotgun sequencing. A high-quality DNA sample was cut into fragments of about 500 bp using the Covaris S220 (Covaris, Woburn, MA, USA). A genomic library with insert size ~350 bp was constructed following the Illumina DNA sample preparation protocol using NEB Next Ultra DNA Library Prep Kit (NEB, USA). Finally, sequencing was performed to generate 150-bp pairedend reads on the HiSeq X Ten platform (Illumina) at Novogene Bioinformatics Technology (Tianjin, China).
The raw reads generated by Illumina sequencing were preprocessed to remove adapter fragments, ambiguous reads (i.e., reads with more than 10% unidentified nucleotides) and low-quality sequences (reads with > 50% bases having phred quality < 20). Then, de novo assembly was carried out using Celera Assembler 8.3 with default settings (Denisov et al., 2008). The scaffolds were used for further microsatellite discovery.
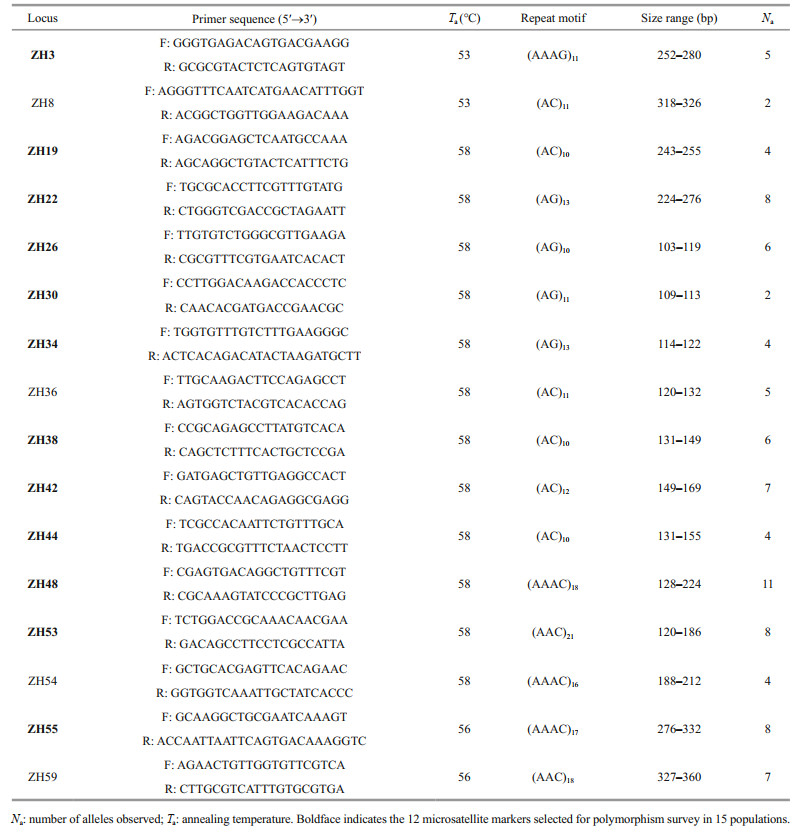
2.3 Microsatellite identification and primer designMicrosatellite discovery and primer design were performed with QDD 3.1.2 (Meglécz et al., 2010) in a local galaxy platform. Sequences containing microsatellites which were defined as pure tandem repeats of di- to hexa-nucleotide motif with at least five uninterrupted repeats, and with 200 bases of flanking regions on both sides were detected and used for primer design in Primer3 (Rozen and Skaletsky, 2000). Primer parameters were set as follows: length from 18 bp to 27 bp with 20 bp as the optimum length; melting temperature between 57 ℃ and 63 ℃ with 60 ℃ as the optimum, and PCR product size between 90 bp and 320 bp. In order to improve the success rate, only primers and loci that passed the following criteria were retained: one primer pair for each locus; only in design category A (target regions without multiple microsatellites, nanosatellite, and homopolymers); and alignment score below 10.
2.4 Validation of microsatellitesPolymorphism analyses were performed on a total of 60 pairs of primers, which were randomly selected out of the 1 636 retained pairs of primers. We used four individuals from QD (Taiping Bay, Qingdao, Shandong Province) and two individuals from ZJT (Zhangjiatai, Rizhao, Shandong Province) to test whether the primers could be PCR amplified with regular primers or not. The successfully amplified primers were then selected for M13-tailed primer method PCR. The cost-effective M13-tailed primer genotyping protocol (Schuelke, 2000) was then adopted to check the polymorphism of microsatellites. The primer mix contains a forward primer with an M13-tail (5′-GGAAACAGCTATGACCATG-3′) on the 5′ end, a universal fluorescent-labelled M13-tail and a reverse primer. A 10-μL PCR amplification volume was adopted, which contains 10-ng genomic DNA, 5-μL 2× PCRmix (Dongsheng Biotech Co., China), 0.025-μmol/L M13-forward primer, 0.25-μmol/L reverse primer, 0.25-μmol/L M13 primer labeled with different fluorescent dyes (FAM, HEX, TAMRA, ROX), and 3.25-μL ultrapure water. Amplifications were performed with the following cycling conditions: initial activation step at 94 ℃ for 4 min, 30 cycles consisting of denaturation for 30 s at 94 ℃, 40 s of gradient annealing at 50–60 ℃, and 40 s of extension at 72℃, followed with 8 cycles consisting of denaturation for 30 s at 94 ℃, 40 s of annealing at 53 ℃, and 40 s of extension at 72 ℃, and a final extension at 72 ℃ for 10 min. The annealing temperature of 53℃ for the eight cycles was based on that of the fluorescent dye-labeled M13(-21) primer (Schuelke, 2000). The PCR products were electrophoresed on a 1.5% agarose gel and only microsatellite loci that produced specific products were sent to Tsingke Biological Technology (Qingdao, China) for capillary electrophoresis using 3730xl DNA Analyzer with the GeneScanTM 500 LIZTM dye Size Standard.
2.5 Microsatellite genotyping, genetic diversity, and population genetic structure analysesAfter the test of validation, 12 microsatellite loci were retained for population genetic analyses. GeneMarker 2.2 (SoftGenetics, State College, USA) was used for allele calling and manual corrections were performed to minimize genotyping errors. Genetic diversity indices including the number of alleles per locus (Na), observed heterozygosity (HO), expected heterozygosity (HE), polymorphism information content (PIC), allelic richness (AR), and inbreeding coefficient (FIS) were calculated using the Excel Microsatellite Toolkit (Park, 2002) and FSTAT (version 2.9.4). Deviations from Hardy-Weinberg equilibrium (HWE) and genotypic linkage disequilibrium (LD) were determined using Genepop 4.5.1 (Rousset, 2008). The significance levels of tests were corrected with standard Bonferroni correction.
Population genetic differentiation was assessed between pairs of populations by calculating pairwise FST and Dest values (two types of population differentiation statistic) with ARLEQUIN 3.5.2 and GenAlEx (Excoffier and Lischer, 2010; Peakall and Smouse, 2012). Global FST was also calculated with Genepop 4.5.1. Then, a multidimensional scaling analysis (MDS) was performed with SPSS 23 (SPSS Inc., Chicago, IL, USA) to visualize patterns of population structuring. Spatial analysis of molecular variance (SAMOVA) was performed in SAMOVA 2.0 (Dupanloup et al., 2002) to define groups of populations that are geographically homogeneous and maximally differentiated from each other, which was run with two to five groups and 1 000 permutations. Bayesian clustering of microsatellite genotypes was performed in STRUCTURE 2.3.4 (Pritchard et al., 2000). Ten replicate runs were performed with the values of K ranging from 2 to 10 (instead of pursuing genetic structure between all of the 15 populations, K is only set to 10 to save time), 1 000 000 Markov Chain Monte Carlo iterations for each run, and a burn-in period of 100 000 iterations with prior knowledge of sampling locations. StructureSelector (Li and Liu, 2018) was used to determine the number of K that best fit the data by calculating Ln Pr(X|K) (Pritchard et al., 2000) and ΔK (Evanno et al., 2005). The software POPTREE 2 (Takezaki et al., 2010) was also used to construct a UPGMA phylogenetic tree based on the DSW distance (Shriver et al., 1995) which takes into account the mutational pattern of microsatellite loci. The number of bootstrap replicates was set at 1 000. Isolation-by-distance (IBD) analyses were conducted for the seven natural rocky populations using IBDWS (Jensen et al., 2005). Google Earth version 4.3 was used to estimate the shoreline geographic distances between sampled populations. Correlation between geographic distance and pairwise FST/(1–FST) were tested with 10 000 randomizations of the data.
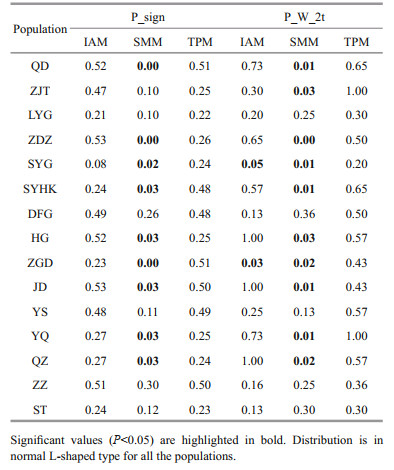
We investigated whether the newly founded populations along Jiangsu Province were affected by genetic drift resulting from bottleneck and/or founder effect. The occurrence of genetic bottleneck was tested with the program Bottleneck 1.2.02 (Piry et al., 1999). Both the Wilcoxon sign-rank test for heterozygosity excess and the sign test that compares the number of loci that present a heterozygosity excess with the number of such loci expected by chance alone was conducted under three mutational models: the infinite alleles model (IAM), the stepwise mutation model (SMM), and the two-phase model (TPM). In the TPM, the default settings of 30% variation from the IAM model and 70% from the SMM model were adopted. Furthermore, effective population size for each population was estimated with NeEstimator v2 by using the linkage disequilibrium method (Do et al., 2014).
3 RESULT 3.1 De novo assembly and microsatellite isolationIn total, 50 579 079 paired-end reads (15.17-Gb data) were obtained. After quality filtering and de novo assembly, 254 132 scaffolds and 259 738 contigs were successfully assembled. The mean bases in scaffolds were 2 045 bp with an N50 of 2 137 bp, and the GC content was 42.29%. Scaffolds were then used for microsatellites detection. A total of 132 044 sequences possessing microsatellite motifs were identified and 103 971 pairs of primers were successfully designed. After filtering, 1 636 primer pairs were retained (Supplementary File S1). The most common repeat motifs of microsatellites were dinucleotide (67.91%), followed by trinucleotide (22.68%), tetranucleotide (8.31%), pentanucleotide(0.86%), and hexanucleotide (0.24%).
Among the 60 primer pairs used for primer testing, 33 were successfully amplified by PCR (Supplementary File S2), of which 16 were successfully amplified microsatellites, showing specific and polymorphic amplification products using labelled M13-tailed primer approach (Table 2). According to the preliminary validation results, we randomly selected 12 loci for subsequent polymorphism survey in the 351 individuals from 15 populations.
|
The 12 microsatellite loci displayed a high level of polymorphisms. Significant deviation from HWE was detected in three loci in all populations (ZH30, ZH34, ZH53), and no LD was detected. These three loci were removed and we retained 9 loci for downstream population genetic analyses (Supplementary File S3). Among populations, the average number of alleles per locus per population (Na) ranged from 9.40 to 12.70. The average expected (HE) and observed (HO) heterozygosity across loci ranged from 0.75 to 0.81 and from 0.44 to 0.64, respectively. The average PIC ranged from 0.71 to 0.77. FIS in all 15 populations were positive and ranged from 0.21 to 0.43 (Table 3).

|
For comparison purposes, the 15 populations were grouped as north group (QD, ZJT), central group (LYG, ZDZ, SYG, SYHK, ZGD, HG, DFG, and JD populations located along Jiangsu Province, north of the Changjiang River estuary) and south group (YS, YQ, QZ, ZZ, and ST populations located south of the Changjiang River estuary) (see Table 1). The global FST was 0.01 and pairwise FST ranged from -0.005 to 0.035 and Dest values ranged from -0.050 to 0.099. Populations that are more geographically distant tended to show higher and significant FST and Dest values, especially those between natural intertidal populations from north and south of the Changjiang River estuary (Table 4). The FST between the north group and central group, north group and south group, central group and south group are 0.014 3, 0.009 8, and 0.001 2, respectively. FST and Dest between the north group and populations from south of the Changjiang River estuary (south group) were generally low but significant. The pairwise FST and Dest within north group was not significant (P > 0.05). The FST values between five populations from the central group (SYHK, DFG, HG, ZGD, and JD) and the south group were less than 0.02 and not statistically significant, which were much lower than those between population from north group and south group. Meanwhile, the Dest values between the central group and the south group also showed a significant decrease. The MDS plots showed a trend of population differentiation between the north group and all the other populations (Fig. 1a).
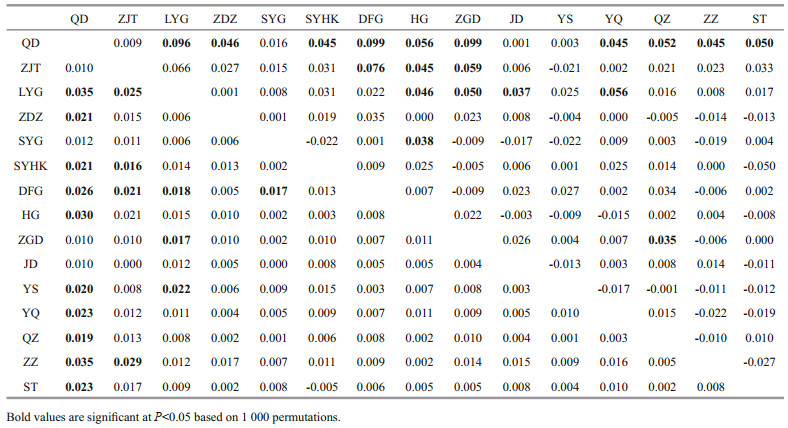
|

|
| Fig.1 Analysis of population structure across 15 geographic populations a. multidimensional scaling analyses (MDS) of FST based on 9 microsatellites markers. Different colors represented three group of populations. Red: natural populations to the south of the Changjiang River; orange: newly formed populations along Jiangsu Province; blue: north natural populations. b. plot of ΔK from StructureSelector. c. STRUCTURE clustering result for K=3. |
The result of STRUCTURE analysis (Fig. 1c) was similar to the results of MDS. The most likely value of K=3 was supported by a peak in values of ΔK (Fig. 1b). But the values of Ln Pr(X|K) showed two plateaus and the peak values at K=10 (Supplementary File S4), which indicated the high genetic polymorphism between different populations. Furthermore, genetic divergence between the newly founded populations in the northern (LYG, ZDZ, SYG, and SYHK) and southern (ZGD, HG, DFG, and JD) parts along Subei Shoal (Central group) was detected. As for the UPGMA tree based on DSW distance, the bootstrap values were extremely low, except the clustering of QD and ZJT (bootstrap: 67)(Fig. 2), which could only indicate that there was a trend of differentiation between the north group (QD, ZJT) and other populations. The genetic and geographic distance matrices were significantly associated in the IBD analysis (R=0.79, P=0.000 6) (Fig. 3).

|
| Fig.2 UPGMA phylogenetic tree based on the DSW distance among 15 populations The blue box represents the north group and the orange box represents the central and south group. |

|
| Fig.3 Plot of pairwise estimates of geographic distance and FST/(1–FST) between populations of L. sinensis |
The results of the two tests of bottleneck effect were consistent (Table 5). In the Sign test, all of the populations were found to be at mutation-drift equilibrium under IAM and TPM. As for the analysis under SMM, the bottleneck effect was found in nine populations. In the Wilcoxon test, significant bottleneck in SYG and ZGD were found under IAM, while a significant bottleneck effect found was found in nine populations under SMM. The estimates of effective population size were infinite for nine populations, five of which were natural populations (Table 3). The six remaining populations showed finite Ne, and four of them were along the coast of Jiangsu Province. However, the upper bound of the confidence interval in all of the populations was infinity. Small sample size or sampling error can lead to infinite estimates of Ne (Do et al., 2014).
The NGS approach has been widely used for developing microsatellite markers in ecological and evolutionary important non-model species including plants, mammals, and mollusks (Göl et al., 2017; Liu et al., 2017; Lopez-Marquez et al., 2018). In the present study, we developed abundant candidate microsatellites for the periwinkle L. sinensis in a cost-effective manner by using an NGS-based method. Compared with the traditional microsatellite development approaches, the NGS-based method could enhance the marker development efficiency in marine gastropods and promote evolutionary and ecological studies in mollusks. The microsatellite markers developed here, to our knowledge, were the first reported microsatellite resources in this ecologically and evolutionarily important family, which could facilitate the applications of microsatellites in other species of the family by cross-species amplification (Pérez et al., 2008; Schoebel et al., 2013).
For L. sinensis, the present study provided a plentiful resource of microsatellites. A total of 1 636 primer pairs were designed and retained after quality control. We successfully amplified 33 of 60 (55.00%) microsatellite loci and 16 (26.67%) worked for the M13-tailed primer method, which could reduce the cost of fluorescent primer labelling (Hayden et al., 2008). However, the amplification success rate was not very high compared with studies that used similar approaches (Xue et al., 2017; Zhang et al., 2019). For species with large population sizes including mollusks, genomic features including unstable flanking sequences and occurrences of cryptic repetitive DNA could be responsible for PCR interference or inconsistencies (McInerney et al., 2011). Although challenges in assembling the draft genome of L. sinensis included high levels of polymorphism and abundant repetitive sequences as in many marine invertebrates (Zhang et al., 2012, 2019), our results still reinforced that NGS is an important and efficient tool for developing genome-wide microsatellites.
The tested loci have a relatively high value for PIC (0.65). Even though significant deviation from the HWE was observed in three out of the 12 selected loci caused by heterozygote deficiency, and the expected heterozygosity was higher than the observed heterozygosity. FIS was positive and very high, which indicated a heterozygosity deficiency that may result from the presence of null alleles and/or inbreeding. Obvious heterozygosity deficiency might result from the presence of null alleles, inbreeding or the Wahlund effect (Andrade et al., 2005; Plutchak et al., 2006; Hui et al., 2017). Deviations from HWE and the presence of a large number of null alleles were also observed in other marine gastropods including Littorina saxatilis(Panova et al., 2008), Littorina scutulata (Holborn et al., 2015), Gibbula divaricate (Lopez-Marquez et al., 2016) and many other marine mollusks including the pacific oyster Crassostrea gigas (Hedgecock et al., 2004) and the Wedge Clam Donax trunculus (Rico et al., 2017), which seems to be common in marine invertebrates (Panova et al., 2008). As demonstrated in other studies, deviations from HWE can be an indicator of various biological processes such as self-fertilization, allele scoring bias, aneuploidy, parental imprinting, spatial and temporal Wahlund effects, age-effects, and direct natural selection on the marker loci (Plutchak et al., 2006). Null alleles at microsatellite loci could have limitations on the estimate of population differentiation (Rico et al., 2017). Null alleles are found in a wide range of taxa (Dakin and Avise, 2004), but it is particularly common in populations with high effective population sizes (Chapuis and Estoup, 2007), including mollusks. Simulation studies and empirical data sets showed that null alleles with frequencies between 5% and 8% should have only minor effects on classical estimates of population differentiation, although higher null allele frequencies may inflate FST and genetic distances (Chapuis and Estoup, 2007). Microsatellites are widely and routinely used as molecular markers in diverse genetic studies, the effectiveness of microsatellites, particularly for L. sinensis, needs further validation and the results should be interpreted cautiously.
4.2 Population genetic differentiation and structureBoth the results of FST and Dest and MDS plots supported genetic differentiation between populations in the north group and the other populations, which was consistent with results of the previous study based on mitochondrial COI sequences (Dong et al., 2016), suggesting significant differentiation between the north group and populations south of Lianyungang, Jiangsu Province. Similar patterns have been observed in marine fishes, mollusks, and crustaceans (Ni et al., 2014), where the phylogeographic structure splits could have been induced by historical glaciation in the Northwest Pacific. During the Last Glacial Maximum, L. sinensis populations might have been isolated in two different marginal seas when most of the East China Sea was exposed (Wang, 1999), and hence, two distinct lineages diverged in that time. However, considering the result of STRUCTURE analysis and the low bootstrap values in the phylogenetic tree, the results may be inconclusive and therefore the conclusion should be taken with caution.
For COI data, the dominant haplotypes were shared by both the north and south populations (Dong et al., 2016). In the present study, the population genetic structure revealed by FST, Dest, STRUCTURE analysis and phylogenetic clustering of populations was weak, which implied high gene flow among populations of L. sinensis. The estimated planktonic larval duration of L. sinensis was 3–10 weeks and the pelagic larvae could be transported by ocean currents to considerable distance during the long pelagic period (Becker et al., 2007). Additionally, many marine invertebrates are able to raft on buoyant macroalgae, which could enhance dispersal over long distances (Nikula et al., 2011). The long planktonic dispersal phases may promote the high population connectivity and maintain weak genetic structure among populations of L. sinensis.
4.3 New populations on the artificial structuresBoth the long planktonic larval stage and new habitat could play important role in structuring populations of L. sinensis. Along the coast of Jiangsu Province, recently constructed artificial structures have provided suitable habitat for intertidal species and L. sinensis was the pioneer species (Dong et al., 2016). SAMOVA analysis showed the maximized genetic differences among groups (FCT=0.034 87; P=0.000) was found when populations were divided into three groups (group 1: QD, ZJT; group 2: LYG, ZDZ, SYG, SYHK, ZGD, HG, DFG, JD, YS, YQ, QZ, ZZ; group 3: ST). The results of genetic distance and the clustering analyses indicated that these newly founded populations (central group) were closely related to the natural populations south of the Changjiang River estuary. Consistent with the conclusion in Dong et al. (2016), these newly founded populations demonstrated the northward spread of populations south of the Changjiang River estuary, which could be accomplished by larval dispersal. Our result suggests that the Changjiang River diluted water did not serve as a barrier for the northward shift of L. sinensis, which was different from previous research suggested (Wang et al., 2015). The coastal current in the Yellow Sea and the East China Sea could have played an important role in the dispersal of planktonic larvae. The energy of flow regime from south to the north could transport the planktonic larval of L. sinensis and enhance the population connectivity across the Changjiang River estuary. Moreover, the small effective population sizes, the bottleneck effect, and the low genetic diversity in some populations on the artificial structures were possibly the results of the founder effect resulting from the colonization of the new habitats. In addition, the significant bottleneck in QD and ZJT under the SMM model might represent characteristics of peripheral populations (Vucetich and Waite, 2003; Göstring et al., 2010).
Little is known of reproductive patterns of L. sinensis, but according to the research of Littoraria pallescens and Littoraria melanostoma (Ng and Williams, 2012), we speculated that this species spawn frequently or rapidly returns to reproductive condition after spawning. Therefore, the overlap between generations could be common. The population sizes in our study were small and the linkage disequilibrium method that we used assumes discrete nonoverlapping generations (Do et al., 2014), therefore, the precision of the Ne estimates may be reduced by overlapping generations, sample size (Antao et al., 2011), and age structure (Robinson and Moyer, 2013). For wildlife populations, it is much difficult to accurately estimate the effective population size owing to the lack of relevant information (Wright et al., 2021). Although the accuracy of population size estimation is uncertain, Ne is recommended primarily as a metric to detect changes over time (Pierson et al., 2018). The strong genetic drift effect in the newly colonized populations with short history could underestimate Ne (Wang and Whitlock, 2003).
Newly-formed populations could provide new sources of larvae, create new dispersal pathways and enhance gene flow (Bulleri and Airoldi, 2005; Glasby et al., 2007; Adams et al., 2014). Results of the serpulid Pomatoceros triqueter and the limpet Patella caerulea have shown that newly formed hard-bottom populations on artificial structures can affect the genetic composition of natural populations (Fauvelot et al., 2009, 2012). Our results show that some populations in the central group were not significantly differentiated from natural populations in both north and south of the Changjiang River estuary. It is thus necessary to investigate the exact contribution of north and south natural populations to the formations of these newly formed populations using high-resolution genetic markers, such as genome-wide genetic markers (Sandoval-Castillo et al., 2018; Cornwell, 2020). However, marker selection should be made with more caution, as putative selected SNP marker would not be efficient for detecting the phylogeographic structure and the founder effects.
5 CONCLUSIONIn conclusion, the present study provided abundant microsatellite markers and genotyped 15 populations of L. sinensis using nine newly characterized microsatellites. This NGS-based microsatellite development approach is a powerful technique for developing microsatellite markers in non-model species. Low genetic differentiation was detected between north and south populations, which suggested high genetic connectivity. Furthermore, populations on artificial structures located along Jiangsu Province showed a close genetic affinity with natural populations south of the Changjiang River estuary, suggesting their southern origin. To our knowledge, we provided here the first microsatellites database and population genetic analysis of L. sinensis from almost the whole distribution range. This study can potentially enhance the understanding of population connectivity in the northwestern Pacific and provide insight into the ecological and evolutionary study of Littoraria.
6 DATA AVAILABILITY STATEMENTThe raw whole genome shotgun sequencing data were deposited in NCBI SRA data set: SRR13107012. And the de novo assembly sequences are accessible via FigShare through https://figshare.com/s/7dcc021c5b25e3ebffd7. Other data generated or analysed during this study are included in this published article and its supplementary information files.
Electronic supplementary materialSupplementary material (Supplementary Files S1–S4) is available in the online version of this article at https://doi.org/10.1007/s00343-021-1079-9
Adams T P, Miller R G, Aleynik D, Burrows M T. 2014. Offshore marine renewable energy devices as stepping stones across biogeographical boundaries. Journal of Applied Ecology, 51(2): 330-338.
DOI:10.1111/1365-2664.12207 |
Andrade S C S, Medeiros H F, Solferini V N. 2005. Homogeneity test of hardy-Weinberg deviations in Brazilian littorinids: evidence for selection?. Journal of Molluscan Studies, 71(2): 167-174.
DOI:10.1093/mollus/eyi019 |
Antao T, Pérez-Figueroa A, Luikart G. 2011. Early detection of population declines: high power of genetic monitoring using effective population size estimators. Evolutionary Applications, 4(1): 144-154.
DOI:10.1111/j.1752-4571.2010.00150.x |
Apolinário M, Coutinho R, Baeta-Neves M H. 1999. Periwinkle (Gastropoda: Littorinidae) habitat selection and its impact upon microalgal populations. Revista Brasileira de Biologia, 59(2): 211-218.
DOI:10.1590/S0034-71081999000200005 |
Becker B J, Levin L A, Fodrie F J, McMillan P A. 2007. Complex larval connectivity patterns among marine invertebrate populations. Proceedings of the National Academy of Sciences of the United States of America, 104(9): 3267-3272.
DOI:10.1073/pnas.0611651104 |
Bulleri F, Airoldi L. 2005. Artificial marine structures facilitate the spread of a non-indigenous green alga, Codium fragile ssp. tomentosoides, in the north Adriatic Sea. Journal of Applied Ecology, 42(6): 1063-1072.
DOI:10.1111/j.1365-2664.2005.01096.x |
Carvalho J, Pereira C, Sotelo G, Costa D, Galindo J, Faria R. 2015. De novo isolation of 17 microsatellite loci for flat periwinkles (Littorina fabalis and L. obtusata) and their application for species discrimination and hybridization studies. Journal of Molluscan Studies, 81(3): 421-425.
DOI:10.1093/mollus/eyv014 |
Chapuis M P, Estoup A. 2007. Microsatellite null alleles and estimation of population differentiation. Molecular Biology and Evolution, 24(3): 621-631.
DOI:10.1093/molbev/msl191 |
Cornwell B H. 2020. Gene flow in the anemone Anthopleura elegantissima limits signatures of local adaptation across an extensive geographic range. Molecular Ecology, 29(14): 2550-2566.
DOI:10.1111/mec.15506 |
Cowen R K, Gawarkiewicz G, Pineda J, Thorrold S R, Werner F E. 2007. Population connectivity in marine systems: an overview. Oceanography, 20(3): 14-21.
DOI:10.5670/oceanog.2007.26 |
Cowen R K, Sponaugle S. 2009. Larval dispersal and marine population connectivity. Annual Review of Marine Science, 1: 443-466.
DOI:10.1146/annurev.marine.010908.163757 |
Cubit J D. 1984. Herbivory and the seasonal abundance of algae on a high intertidal rocky shore. Ecology, 65(6): 1904-1917.
DOI:10.2307/1937788 |
Dakin E E, Avise J C. 2004. Microsatellite null alleles in parentage analysis. Heredity, 93(5): 504-509.
DOI:10.1038/sj.hdy.6800545 |
Denisov G, Walenz B, Halpern A L, Miller J, Miller N, Levy S, Sutton G. 2008. Consensus generation and variant detection by Celera Assembler. Bioinformatics, 24(8): 1035-1040.
DOI:10.1093/bioinformatics/btn074 |
Do C, Waples R S, Peel D, Macbeth G M, Tillett B J, Ovenden J R. 2014. NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Molecular Ecology Resources, 14(1): 209-214.
DOI:10.1111/1755-0998.12157 |
Dong Y W, Huang X W, Wang W, Li Y, Wang J. 2016. The marine 'Great Wall' of China: local- and broad-scale ecological impacts of coastal infrastructure on intertidal macrobenthic communities. Diversity and Distributions, 22(7): 731-744.
DOI:10.1111/ddi.12443 |
Doropoulos C, Roff G, Bozec Y M, Zupan M, Werminghausen J, Mumby P J. 2016. Characterizing the ecological tradeoffs throughout the early ontogeny of coral recruitment. Ecological Monographs, 86(1): 20-44.
DOI:10.1890/15-0668.1 |
Dupanloup I, Schneider S, Excoffier L. 2002. A simulated annealing approach to define the genetic structure of populations. Molecular Ecology, 11(12): 2571-2581.
DOI:10.1046/j.1365-294X.2002.01650.x |
Estoup A, Presa P, Krieg F, Vaiman D, Guyomard R. 1993. (CT)n and (GT)n microsatellites: a new class of genetic markers for Salmo trutta L. (brown trout). Heredity, 71(5): 488-496.
DOI:10.1038/hdy.1993.167 |
Evanno G, Regnaut S, Goudet J. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology, 14(8): 2611-2620.
DOI:10.1111/j.1365-294X.2005.02553.x |
Excoffier L, Lischer H E L. 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3): 564-567.
DOI:10.1111/j.1755-0998.2010.02847.x |
Fauvelot C, Bertozzi F, Costantini F, Airoldi L, Abbiati M. 2009. Lower genetic diversity in the limpet Patella caerulea on urban coastal structures compared to natural rocky habitats. Marine Biology, 156(11): 2313-2323.
DOI:10.1007/s00227-009-1259-1 |
Fauvelot C, Costantini F, Virgilio M, Abbiati M. 2012. Do artificial structures alter marine invertebrate genetic makeup?. Marine Biology, 159(12): 2797-2807.
DOI:10.1007/s00227-012-2040-4 |
Flanagan S P, Jones A G. 2019. The future of parentage analysis: from microsatellites to SNPs and beyond. Molecular Ecology, 28(3): 544-567.
DOI:10.1111/mec.14988 |
Glasby T M, Connell S D, Holloway M G, Hewitt C L. 2007. Nonindigenous biota on artificial structures: could habitat creation facilitate biological invasions?. Marine Biology, 151(3): 887-895.
DOI:10.1007/s00227-006-0552-5 |
Göl S, Göktay M, Allmer J, Doğanlar J, Frary A. 2017. Newly developed SSR markers reveal genetic diversity and geographical clustering in spinach (Spinacia oleracea). Molecular Genetics and Genomics, 292(4): 847-855.
DOI:10.1007/s00438-017-1314-4 |
Göstring L, Chew M T, Orlova A, Höidén-Guthenberg I, Wennborg A, Carlsson J, Frejd F Y. 2010. Quantification of internalization of EGFR-binding Affibody molecules: methodological aspects. International Journal of Oncology, 36(4): 757-763.
|
Hayden M J, Nguyen T M, Waterman A, Chalmers K J. 2008. Multiplex-ready PCR: a new method for multiplexed SSR and SNP genotyping. BMC Genomics, 9: 80.
DOI:10.1186/1471-2164-9-80 |
Hedgecock D, Li G, Hubert S, Bucklin K, Ribes V. 2004. Widespread null alleles and poor cross-species amplification of microsatellite DNA loci cloned from the Pacific oyster, Crassostrea gigas. Journal of Shellfish Research, 23(2): 379-385.
|
Holborn M K, Krick M V, Pedersen S, Hunt D A G A, Boulding E G. 2015. Polymorphic microsatellite loci for Littorina plena show no population structure between the eastern and western coasts of Vancouver Island, Canada. Journal of Molluscan Studies, 81(3): 407-411.
DOI:10.1093/mollus/eyv002 |
Hui M, Nuryanto A, Kochzius M. 2017. Concordance of microsatellite and mitochondrial DNA markers in detecting genetic population structure in the boring giant clam Tridacna crocea across the Indo-Malay Archipelago. Marine Ecology, 38(1): e12389.
DOI:10.1111/maec.12389 |
Jensen J L, Bohonak A J, Kelley S T. 2005. Isolation by distance, web service. BMC Genetics, 6: 13.
|
Kennington W J, Lukehurst S S, Johnson M S. 2008. Characterization of microsatellite loci for the littorine snail Bembicium vittatum. Molecular Ecology Resources, 8(6): 1463-1465.
DOI:10.1111/j.1755-0998.2008.02247.x |
Li Y L, Liu J X. 2018. StructureSelector: a web-based software to select and visualize the optimal number of clusters using multiple methods. Molecular Ecology Resources, 18(1): 176-177.
DOI:10.1111/1755-0998.12719 |
Liu S X, Hou W, Sun T L, Xu Y T, Li P, Yue B S, Fan Z X, Li J. 2017. Genome-wide mining and comparative analysis of microsatellites in three macaque species. Molecular Genetics and Genomics, 292(3): 537-550.
DOI:10.1007/s00438-017-1289-1 |
Lopez-Marquez V, Garcia-Jimenez R, Calvo M, Templado J, Machordom A. 2018. Isolation of microsatellite loci for the endangered vermetid gastropod Dendropoma lebeche using Illumina MiSeq next generation sequencing technology. Molecular Biology Reports, 45(6): 2775-2781.
DOI:10.1007/s11033-018-4346-x |
Lopez-Marquez V, Garcia-Jimenez R, Templado J, Machordom A. 2016. Development and characterization of 26 novel microsatellite loci for the trochid gastropod Gibbula divaricata (Linnaeus, 1758), using Illumina MiSeq next generation sequencing technology. PeerJ, 4: e1789.
DOI:10.7717/peerj.1789 |
McInerney C E, Allcock A L, Johnson M P, Bailie D A, Prodöhl P A. 2011. Comparative genomic analysis reveals speciesdependent complexities that explain difficulties with microsatellite marker development in molluscs. Heredity, 106(1): 78-87.
DOI:10.1038/hdy.2010.36 |
Meglécz E, Costedoat C, Dubut V, Gilles A, Malausa T, Pech N, Martin J F. 2010. QDD: a user-friendly program to select microsatellite markers and design primers from large sequencing projects. Bioinformatics, 26(3): 403-404.
DOI:10.1093/bioinformatics/btp670 |
Menge B A. 2000. Top-down and bottom-up community regulation in marine rocky intertidal habitats. Journal of Experimental Marine Biology and Ecology, 250(1-2): 257-289.
DOI:10.1016/S0022-0981(00)00200-8 |
Ng T P T, Williams G A. 2012. Contrasting reproductive traits in two species of mangrove-dwelling littorinid snails in a seasonal tropical habitat. Invertebrate Biology, 131(3): 177-186.
DOI:10.1111/j.1744-7410.2012.00269.x |
Ni G, Li Q, Kong L F, Yu H. 2014. Comparative phylogeography in marginal seas of the northwestern Pacific. Molecular Ecology, 23(3): 534-548.
DOI:10.1111/mec.12620 |
Nikula R, Spencer H G, Waters J M. 2011. Comparison of population-genetic structuring in congeneric kelp-versus rock-associated snails: a test of a dispersal-by-rafting hypothesis. Ecology and Evolution, 1(2): 169-180.
DOI:10.1002/ece3.16 |
Panova M, Makinen T, Fokin M, André C, Johannesson K. 2008. Microsatellite cross-species amplification in the genus Littorina and detection of null alleles in Littorina saxatilis. Journal of Molluscan Studies, 74(2): 111-117.
DOI:10.1093/mollus/eym052 |
Park S D E. 2002. Trypanotolerace in West African Cattle and the Population Genetic Effects Of Selection. University of Dublin, Dublin.
|
Peakall R, Smouse P E. 2012. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics, 28(19): 2537-2539.
DOI:10.1093/bioinformatics/bts460 |
Pérez M, Guinez R, Llavona A, Toro J E, Astorga M, Presa P. 2008. PERMANENT GENETIC RESOURCES: Development of microsatellite markers for the ecosystem bioengineer mussel Perumytilus purpuratus and cross-priming testing in six Mytilinae genera. Molecular Ecology Resources, 8(2): 449-451.
DOI:10.1111/j.1471-8286.2007.01989.x |
Pierson J C, Graves T A, Banks S C, Kendall K C, Lindenmayer D B. 2018. Relationship between effective and demographic population size in continuously distributed populations. Evolutionary Applications, 11(7): 1162-1175.
DOI:10.1111/eva.12636 |
Piry S, Luikart G, Cornuet J M. 1999. Computer note.BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. Journal of Heredity, 90(4): 502-503.
DOI:10.1093/jhered/90.4.502 |
Plutchak L L, Simmons R E, Woodruff D S. 2006. Multilocus allozyme heterozygote deficiencies in Crepidula onyx: geographic and temporal patterns among adult snails in Mission Bay, California. Journal of Molluscan Studies, 72(4): 337-348.
DOI:10.1093/mollus/eyl013 |
Pritchard J K, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics, 155(2): 945-959.
DOI:10.1093/genetics/155.2.945 |
Raffaelli D, Paine R T. 1995. Marine rocky shores and community ecology: an experimentalist′s perspective. Journal of Animal Ecology, 64(3): 425-427.
|
Reid D G, Dyal P, Williams S T. 2010. Global diversification of mangrove fauna: a molecular phylogeny of Littoraria (Gastropoda: Littorinidae). Molecular Phylogenetics and Evolution, 55(1): 185-201.
DOI:10.1016/j.ympev.2009.09.036 |
Reid D G. 1989. The comparative morphology, phylogeny and evolution of the gastropod family Littorinidae. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 324(1220): 1-110.
|
Reid D G. 2001. New data on the taxonomy and distribution of the genus Littoraria Griffith and Pidgeon, 1834 (Gastropoda: Littorinidae) in Indo-West Pacific mangrove forests. Nautilus, 115(4): 115-139.
|
Rico C, Cuesta J A, Drake P, Macpherson E, Bernatchez L, Marie A D. 2017. Null alleles are ubiquitous at microsatellite loci in the Wedge Clam (Donax trunculus). PeerJ, 5: e3188.
DOI:10.7717/peerj.3188 |
Robinson J D, Moyer G R. 2013. Linkage disequilibrium and effective population size when generations overlap. Evolutionary Applications, 6(2): 290-302.
DOI:10.1111/j.1752-4571.2012.00289.x |
Rosa A L, Lima-Rezende C A, Rodrigues F P, Caparroz R. 2019. Development and characterization of 19 polymorphic microsatellite loci from the red-cowled cardinal (Paroaria dominicana, Passeriformes, Aves) using next-generation sequencing. Molecular Biology Reports, 46(5): 5531-5536.
DOI:10.1007/s11033-019-04919-z |
Rousset F. 2008. GENEPOP'007: a complete reimplementation of the GENEPOP software for Windows and Linux. Molecular Ecology Resources, 8(1): 103-106.
DOI:10.1111/j.1471-8286.2007.01931.x |
Rozen S, Skaletsky H. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods in Molecular Biology, 132: 365-386.
|
Sandoval-Castillo J, Robinson N A, Hart A M, Strain L W S, Beheregaray L B. 2018. Seascape genomics reveals adaptive divergence in a connected and commercially important mollusc, the greenlip abalone (Haliotis laevigata), along a longitudinal environmental gradient. Molecular Ecology, 27(7): 1603-1620.
DOI:10.1111/mec.14526 |
Schoebel C N, Brodbeck S, Buehler D, Cornejo C, Gajurel J, Hartikainen H, Keller D, Leys M, Říčanová Š, Segelbacher G, Werth S, Csencsics D. 2013. Lessons learned from microsatellite development for nonmodel organisms using 454 pyrosequencing. Journal of Evolutionary Biology, 26(3): 600-611.
DOI:10.1111/jeb.12077 |
Schuelke M. 2000. An economic method for the fluorescent labeling of PCR fragments. Nature Biotechnology, 18(2): 233-234.
DOI:10.1038/72708 |
Selkoe K A, D'aloia C C, Crandall E D, Iacchei M, Liggins L, Puritz J B, von der Heyden S, Toonen R J. 2016. A decade of seascape genetics: contributions to basic and applied marine connectivity. Marine Ecology Progress Series, 554: 1-19.
DOI:10.3354/meps11792 |
Selkoe K A, Toonen R J. 2006. Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecology Letters, 9(5): 615-629.
DOI:10.1111/j.1461-0248.2006.00889.x |
Sharma P C, Grover A, Kahl G. 2007. Mining microsatellites in eukaryotic genomes. Trends in Biotechnology, 25(11): 490-498.
DOI:10.1016/j.tibtech.2007.07.013 |
Shriver M D, Jin L, Boerwinkle E, Deka R, Ferrell R E, Chakraborty R. 1995. A novel measure of genetic distance for highly polymorphic tandem repeat loci. Molecular Biology and Evolution, 12(5): 914-920.
|
Takezaki N, Nei M, Tamura K. 2010. POPTREE2: software for constructing population trees from allele frequency data and computing other population statistics with Windows interface. Molecular Biology and Evolution, 27(4): 747-752.
DOI:10.1093/molbev/msp312 |
Tie A D, Boulding E G, Naish K A. 2000. Polymorphic microsatellite DNA markers for the marine gastropod Littorina subrotundata. Molecular Ecology, 9(1): 108-110.
DOI:10.1046/j.1365-294x.2000.00764-2.x |
Vucetich J A, Waite T A. 2003. Spatial patterns of demography and genetic processes across the species' range: Null hypotheses for landscape conservation genetics. Conservation Genetics, 4(5): 639-645.
DOI:10.1023/A:1025671831349 |
Wang J L, Whitlock M C. 2003. Estimating effective population size and migration rates from genetic samples over space and time. Genetics, 163(1): 429-446.
DOI:10.1093/genetics/163.1.429 |
Wang J, Tsang L M, Dong Y W. 2015. Causations of phylogeographic barrier of some rocky shore species along the Chinese coastline. BMC Evolutionary Biology, 15: 114.
DOI:10.1186/s12862-015-0387-0 |
Wang P X. 1999. Response of Western Pacific marginal seas to glacial cycles: paleoceanographic and sedimentological features. Marine Geology, 156(1-4): 5-39.
DOI:10.1016/S0025-3227(98)00172-8 |
Wright P G R, Schofield H, Mathews F. 2021. Can effective population size estimates be used to monitor population trends of woodland bats?. A case study of Myotis bechsteinii Ecology and Evolution, 11(5): 2015-2023.
|
Xue D X, Li Y L, Liu J X. 2017. A rapid and cost-effective approach for the development of polymorphic microsatellites in non-model species using paired-end RAD sequencing. Molecular Genetics and Genomics, 292(5): 1165-1174.
DOI:10.1007/s00438-017-1337-x |
Zhang G F, Fang X D, Guo X M, Li L, Luo R B, Xu F, Yang P C, Zhang L L, Wang X T, Qi H G, Xiong Z Q, Que H Y, Xie Y L, Holland P W H, Paps J, Zhu Y B, Wu F C, Chen Y X, Wang J F, Peng C F, Meng J, Yang L, Liu J, Wen B, Zhang N, Huang Z Y, Zhu Q H, Feng Y, Mount A, Hedgecock D, Xu Z, Liu Y J, Domazet-Lošo T, Du Y S, Sun X Q, Zhang S D, Liu B H, Cheng P Z, Jiang X T, Li J, Fan D D, Wang W, Fu W J, Wang T, Wang B, Zhang J B, Peng Z Y, Li Y X, Li N, Wang J P, Chen M S, He Y, Tan F J, Song X R, Zheng Q M, Huang R L, Yang H L, Du X D, Chen L, Yang M, Gaffney P M, Wang S, Luo L H, She Z C, Ming Y, Huang W, Zhang S, Huang B Y, Zhang Y, Qu T, Ni P X, Miao G Y, Wang J Y, Wang Q, Steinberg C E W, Wang H Y, Li N, Qian L M, Zhang G J, Li Y R, Yang H M, Liu X, Wang J, Yin Y, Wang J. 2012. The oyster genome reveals stress adaptation and complexity of shell formation. Nature, 490(7418): 49-54.
DOI:10.1038/nature11413 |
Zhang X M, Zhou Y, Li Y L, Liu J X. 2019. Development of microsatellite markers for the seagrass Zostera japonica using next-generation sequencing. Molecular Biology Reports, 46(1): 1335-1341.
DOI:10.1007/s11033-018-4491-2 |