 2021, Vol. 39
2021, Vol. 39Institute of Oceanology, Chinese Academy of Sciences
Article Information
- SUN Qiqi, SONG Jinming, LI Xuegang, YUAN Huamao, WANG Qidong
- The bacterial diversity and community composition altered in the oxygen minimum zone of the Tropical Western Pacific Ocean
- Journal of Oceanology and Limnology, 39(5): 1690-1704
- http://dx.doi.org/10.1007/s00343-021-0370-0
Article History
- Received Oct. 12, 2020
- accepted in principle Dec. 21, 2020
- accepted for publication Jan. 20, 2021




2 Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences, Qingdao 266071, China;
3 Laboratory for Marine Ecology and Environmental Science, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao 266237, China;
4 University of Chinese Academy of Sciences, Beijing 100049, China;
5 Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao 266071, China
Oxygen is an important component in marine ecosystems, which profoundly structure the bacterial and archaeal communities (Aldunate et al., 2018) and mediate the biogeochemical cycles in the open sea (Spietz et al., 2015). Recent modelling results suggested that oxygen will decrease continuously over the next decades to centuries, referred as "ocean deoxygenation". Globally, over a quarter-million square kilometers of marine ecosystems are threatened by low dissolved oxygen levels, or hypoxia (Diaz and Rosenberg, 2008), which can result in exclusion of resident macroorganisms and shift in bacterial diversity and community composition. Marine oxygen minimum zones (OMZs) are intrinsic water column features with low oxygen concentrations (Paulmier and Ruiz-Pino, 2009) and generated primarily due to poor ventilation, sluggish circulation, and a high demand of oxygen by microbial aerobic respiration (Ulloa et al., 2013). The OMZ in the open sea included three layers according to the vertical variation of oxygen levels: upper oxycline (UO, dissolved oxygen (DO) decreased rapidly from the normal level), OMZ core (OM, characterized by the minimum DO level) and lower oxycline (LO, DO recovered from the minimum), with the mixed layer (ML, higher level DO) and suboxycline (SO, DO continue to increase) located above and below the OMZ (Wishner et al., 2013). Understanding the distribution pattern of microbes associated with dissolved oxygen gradients is crucial to forecast potential alterations in bacterial assemblages when confronting global climate change, which is of importance in driving global biogeochemical processes (Azam and Malfatti, 2007).
OMZs could provide an array of niches inhabited by metabolically diverse groups of bacteria such as Thaumarchaeota dominated in dysoxic waters (Gillies et al., 2015) and SUP05 group that dominated the suboxic and anoix waters (Hawley et al., 2014). Generally, the bacterial richness was higher in the OMZs than in the oxic surface and deeper oxycline (Stevens and Ulloa, 2008). Microbial assemblages, in turn, profoundly mediate the development of OMZs and maintain their intensity (Gonsalves et al., 2011). OMZs support unique habitats in which organic matter is mineralized by microorganisms, mostly via anaerobic processes (Jain et al., 2014). Furthermore, the OMZs have long been recognized as biogeochemical hot spots, as energy is increasingly diverted away from high trophic levels into microbial community metabolism as oxygen levels decline, causing changes in carbon and nutrient cycling (Hawley et al., 2014). So far, OMZs have been extensively studied for their role in nitrogen and sulfur cycling within the water column of Arabian Sea (Jayakumar et al., 2009; Gonsalves et al., 2011), the Saanich Inlet on Vancouver Island British Columbia Canada (Hawley et al., 2014), the eastern tropical North Pacific (ETNP) (Carolan et al., 2015) and the India Ocean (Fernandes et al., 2020) etc. Moreover, microorganisms within OMZs mediate loss of fixed nitrogen to the atmosphere through denitrification (Naqvi, 1994) and anaerobic oxidation of ammonia (Dalsgaard et al., 2012), and consequently alter nutrient cycles and the marine carbon budget (Li et al., 2017; Tian et al., 2019). The contribution of the OMZs on the oceanic sources and sinks budget of CO2 has also been researched (Paulmier et al., 2011). As reported, oxygen depletion leads to loss of one third of the reactive nitrogen and production of greenhouse gases (Wright et al., 2012; Tian et al., 2020). Therefore, expansion and intensification of OMZs in the global ocean is of global concern, because of climate change with resulting feedback on marine ecosystem function (Wright et al., 2012). Oxygen depletion changes the diversity, structure, and function of marine microbial communities (Beman and Carolan, 2013; Aldunate et al., 2018). At the community level, microbial diversity presented increasing (Stevens and Ulloa, 2008), decreasing (Bryant et al., 2012), or unimodal pattern (Beman and Carolan, 2013) as oxygen levels decreased with depth. In spite of the global biogeochemical and climatic importance of OMZs, the diversity and distribution of OMZ microorganisms in the tropical areas such as Tropical Western Pacific Ocean (TWPO), have received little attention.
In the seamount area of TWPO, microbial communities within the water column have been known to show well-defined stratified patterns (Sun et al., 2020), yet to what extent oxygen contributes to such pattern remain unclear. In order to understand microbial response pattern under low-oxygen stress and relevant mechanism, bacterial diversity and community composition within the water column were investigated using Illumina sequencing of 16S rRNA V4 regions from 66 water samples collected from the six sampling stations. The predicted functional profiling from the samples were also studied. We hypothesized that the bacterial diversity and community composition do not decrease or increase monotonically with depth, but instead track the variation of DO.
2 MATERIAL AND METHOD 2.1 Site description and samplingThe open sea investigation was carried out in the seamount area of the Tropical Western Pacific Ocean (TWPO: 0°–16°N, 125°E–165°E, the same site from Sun et al. (2020)) by the Institute of Oceanology, Chinese Academy of Sciences in the R/V Kexue (Science in Chinese) cruise in May to June 2019. The seamount M5 (10.1°N, 140.2°E, summit: 810 m) is located in the warm pool region in the TWPO (Hoerling et al., 2001), characterized by highlystratified water throughout the year. Thermocline was observed at the depths from 75 m to 150 m within the water column (Sun et al., 2020), which limits the movement of oxygenated waters (Hoerling et al., 2001; Ma et al., 2019).
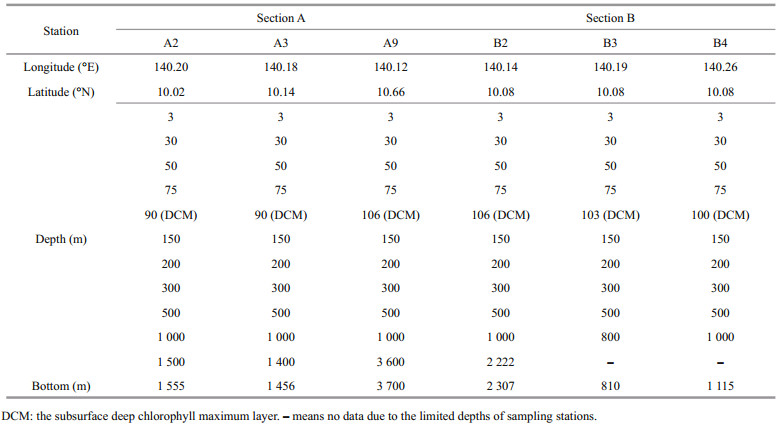
During the cruise, six stations of two sections (A and B) were visited (Fig. 1): stations A2 is located at south slope of M5, A3 is at the north slope of M5, and A9 is seated far away in the open sea; B2 is in the west of M5, B3 is above the summit, and B4 is located at the east slope of M5. Water layers were sampled at certain increment (Table 1). In total, 66 water layers of six stations were sampled using Niskin water samplers (KC-Denmark, Denmark), which were conducted at certain time at night from 10 pm to 6 am so as to avoid day-night cycles and tide currents (Ma et al., 2020). For each layer, 2.0-L seawater sample was filtered through a polycarbonate membrane (0.22-μm pore) and all the filters were stored in DNAfree 5-mL tubes at -80 ℃ for DNA extraction. Dissolved oxygen was measured on board according to the China's national standard of Specification for Oceanographic Survey (GB12763.6-2007): after being collected for each layer with a brown iodine volumetric flask, 50-mL water sample was fixed with manganese sulfate and an alkaline potassium iodide solution and then measured in situ with a Winkler iodometry at relative standard deviation of ≤2% (Zuo et al., 2019). Temperature (T) was measured simultaneously with water sampling using a conductivity-temperature-depth (CTD) profile (SeaBird, SBE911).

|
| Fig.1 Map of investigation stations (a) and the vertical distribution of DO in sections A (b) and B (c) |
According to the manufacturer's instructions, 50-ng genomic DNA was extracted from the filter per layer using a FastDNA Spin Kit (MP Biomedical, Cleveland, OH, USA) with cetyl trimethyl ammonium bromide (CTAB)/solid dodecyl sulfate (SDS) method. The hypervariable V4 region of the 16S rRNA gene from the bacteria were amplified using primers 515F and 806R (Caporaso et al., 2011). Sequencing was performed on a platform of Illumina HiSeq 2500 (Illumina Corporation, San Diego, USA). There were eventually 68 332 high-quality prokaryotic sequences per sample being generated, with average length of 408 bp.
The sequence analysis was conducted using the UPARSE software package with the UPARSE-OTU and UPARSE-OTU ref algorithms and sequences with ≥97% similarity were clustered into operational taxonomic units (OTUs) (Edgar, 2013). Taxonomy was assigned using the Ribosomal Database Project classifier (Wang et al., 2007). Alpha-diversity indices were generated based on the obtained OTUs. The Chao1 richness and Shannon diversity indices were used to estimate species richness and community diversity of microbes. The predictive functional assignments of the bacterial communities of the 66 samples were obtained using the FAPROTAX. The high-throughput sequencing data were uploaded to the NCBI Sequence Read Archive database with accession numbers PRJNA689328.
2.3 Data analysisPrincipal coordinate analysis (PCoA) was conducted to identify the dissimilarity in bacterial community structure among water layers and the Redundancy analysis (RDA) was performed to identify the relationship between bacterial groups and DO, T, both of which were conducted using R software v.4.0.3 (R Core Team, 2013). The line/scatter charts and the stacked horizontal bars were plotted using Sigmaplot software v.12.5 (Systat Software Inc., San José, CA, USA).
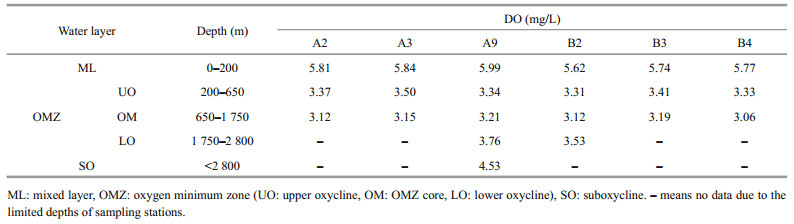
3 RESULT 3.1 OMZ layerThe marine seawater within the water column in the TWPO has DO concentration of 3.01–6.68 mg/L, with oxycline could be clearly observed at both sections (Fig. 2). The core of OMZ could be clearly identified to be situated at 650–1 750 m, with the DO threshold of 3.5 mg/L (Figs. 1 & 2). Therefore, water samples could be artificially divided into five layers according to DO gradients and the oxycline (Wishner et al., 2013): the mixed layer (ML) was located at depths of 0–200 m, the OMZ rested between the depths of 200 m and 2 800 m (with the OMZ core situated at 650–1 750 m), and the SO seated below 2 800 m, if any (Table 2; Fig. 2).

|
| Fig.2 Vertical profiles of DO within water column using CTD data ML: mixed layer; UO: upper oxycline; OM: OMZ core; LO: lower oxycline; SO: suboxycline. |
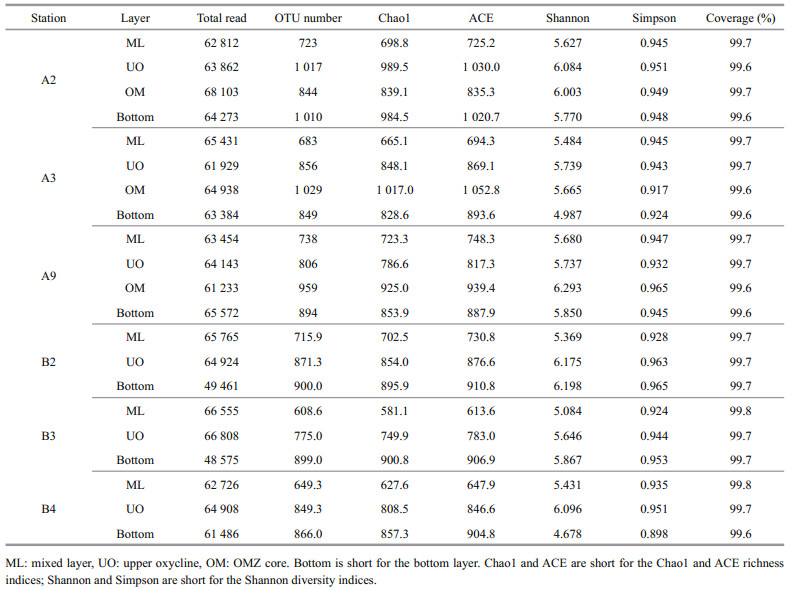
There were in total 4 099 899 high-quality sequences for bacterial 16S rRNA genes of all samples with an average of 68 332 sequences per sample. Eventually, the bacterial community had 59 929 clustered OTUs, with the greatest unique OTUs detected in the OMZs. In this study, the rarefaction curves of all the samples tended to be gentle (Supplementary Fig.S1) with the coverage reaching up to over 99% (Table 3), which indicated that most species had been detected and the dataset used in the subsequent analysis was reliable. Both observed richness (Chao1) and community diversity (Shannon) of bacterial 16S rRNA OTUs fluctuated and generally increased since chlorophyll maximum layer (DCM), with the DO vertically declined (Fig. 3). Overall, bacterial alpha-diversity indices were obviously lower in the ML than in other layers (Table 3), indicating that ocean deoxygenation may promote bacterial alpha-diversity. Indeed, bacterial alphadiversity indices did not increase monotonically with depth within the water column, but instead displayed unimodal pattern (Fig. 3). More specifically, the observed richness tended to increase with depth in ML and peaked at the depths between 150 m and 500 m (Fig. 3a). However, the community diversity tended to increase with depth in ML and peaked on the upper edge of the OMZ and then decreased rapidly (Fig. 3b). Within the OMZ, both the richness and diversity had a slight re-rise at the OMZ core (1 000 m). Notably the bottom layer of each station shared the same bacterial communities, due to the effect of beneath macroorganisms or other factors at the seafloor overwhelmed that of DO (Sun et al., 2020). In this study, only ML, UO, OM, and the bottom layers were observed, due to the limited depths of sampling stations.
|

|
| Fig.3 Vertical profiles of Chao1 richness (a) and Shannon diversity (b) indices within water column |
Proteobacteria, Firmicutes, Cyanobacteria, Actinobacteria, and Thaumarchaeota predominated the water samples across stations, accounting for 88% to 98% of the total reads of 16S rRNA bacteria gene. Overall, bacterial communities transitioned from being Proteobacteria and Cyanobacteria-dominant in the ML to being Proteobacteria and Thaumarchaeotadominant in UO and OM, and then to being Firmicutes and Actinobacteria-dominant in the bottom layer, regardless of stations. At the class level, bacterial communities generally transitioned from being Alphaproteobacteria and Gammaproteobacteriadominant in ML to being Gammaproteobacteria and Nitrososphaeria/Deltaproteobacteria-dominant in UO and OM, and then to being Clostridia and unidentified_ Actinobacteria-dominant in the bottom layer, for most stations (Fig. 4). It was notable that the bacterial community composition started to change since the DCM (Supplementary Fig.S2), at which DO decline with depths (Sun et al., 2020). However, the predominant taxa shifted at 200 m with the DO threshold of 4.5 mg/L.

|
| Fig.4 Vertical profiles of relative abundances of bacterial communities at the class level a. A2; b. A3; c. A9; d. B2; e. B3; f. B4. ML: mixed layer, UO: upper oxycline; OM: OMZ core. Bottom is short for the bottom layer. |
At the order level, bacterial communities changed from being unidentified_Alphaproteobacteriadominant in ML to being Nitrosopumilales and Alteromonadales-dominant in UO and OM, and then turned to being Clostridiales and Bififobacterialesdominant at the bottom layer, for most stations. At the family level, bacterial communities changed from being unidentified_Alphaproteobacteria-dominant in ML to being Nitrosopumilaceae and unidentified_ Deltaproteobacteria/Alteromonadaceae-dominant in UO, and then turned to being Bifidobacteriaceae or Lachnospiraceae-dominant in the bottom layer. Especially, the OM was predominated by unidentified_ Cardiobacteriales at A2, Alteromonadaceae at A3 and Nitrosopumilaceae at A9.
PCoA results show that 46.91% and 34.77% of the spatial variations of bacterial communities could be explained by the first and second axes (P < 0.05), indicating heterogeneous distribution of the bacterial assemblages (Fig. 5a). The bacterial communities of ML and UO clustered and separated from each other, while those of LO and OM clustered but mixed, indicating that the bacterial community structure within water column was extremely shaped by DO gradients. DO and T were selected to conduct RDA of the bacteria, as DO and T decrease in parallel in the ocean (Beman and Carolan, 2013). Within water column, DO (R2=0.82, P < 0.001) and T (R2=0.82, P < 0.001) could explain 41.4% of the variation of bacterial communities among layers (P < 0.001), with 37.02% and 4.38% of the total variation could be explained by the first two axes. DO, T were mainly loaded on the first axis, and DO was slightly loaded on the second axis (Fig. 5b). As far as DO was solely concerned, 40.37% of the variation of bacterial communities among layers could be explained (P < 0.001), with the first two axes explaining 32.78% and 14.04% of the total variation (Supplementary Fig.S3).

|
| Fig.5 PCoA (a) and bi-plot of RDA (b) of bacterial community composition in the different layers ML: mixed layer, UO: upper oxycline, OM: OMZ core. Bottom is short for the bottom layer. |
FAPROTAX function annotation results showed that the predicted OTUs presented significant variation within water column and extremely shaped by DO (Fig. 6, Supplementary Fig.S4). For most stations, the water column was generally predominant by bacterial functional OTUs indicating chemoheterotrophy, cyanobacteria and photoautotrophy above 75 m, by OTUs indicating (aerobic) chemoheterotrophy, and then OTUs indicating nitrogen/sulfide cycling between the DCM and 1 000 m, and by OTUs indicating chemoheteotrophy and fermentation at the seafloor (Supplementary Fig.S4). When divided by DO gradients, water layers changed from being dominated by the bacterial functional OTUs indicating cyanobacteria and photoautotrophy in ML, to being dominated by the bacterial functional OTUs indicating (aerobic) chemoheterotrophy, and then OTUs indicating nitrogen/sulfide cycling in UO and OM. However, the bottom layers were predominated by the OTUs indicating chemoheterotrophy and fermentation (Fig. 6). It was notable that the OMZ (especially the UO) had the greatest OTUs indicating nitrate/sulfide transformation (Supplementary Fig.S5), which peaked at 200 m or 300 m and decreased with depths until the core of OMZs (1 000 m).

|
| Fig.6 Relative abundances of FAPROTAX function annotation for the bacteria from the water layers a. A2; b. A3; c. A9; d. B2; e. B3; f. B4. ML: mixed layer; UO: upper oxycline; OM: OMZ core. Bottom is short for the bottom layer. |
As a subsequent research, the present study focused on the DO-associated distribution pattern of bacterial diversity, community composition, and the predicted function within water column of the TWPO, especially that of OMZ. However, both the seamount-related horizontal variability of bacterial properties and the uniqueness of individual stations were excluded, which have been reported in our previous study (Sun et al., 2020).
4.1 Unimodal pattern of bacterial alpha-diversity with DOOur data provided evidence that ocean deoxygenation may promote bacterial alpha-diversity, which were generally higher in the OMZ than in ML (Fig. 3). Actually, the bacterial alpha-diversity indices did not increase monotonically with DO but instead presented unimodal pattern, with tipping point on the boundary region between ML and OMZ, especially in UO (Fig. 3). To clarify the unimodal pattern, the dataset of individual stations was pooled to develop their relationship with DO (Fig. 7, n=66), and the richness and diversity fitted divergent and segmental functions (Fig. 7). More specifically, the richness increased extremely with decreasing DO when it was higher than 5.3 mg/L, and remained high values and showed negligible variation when it was lower than 5.3 mg/L (Fig. 7a), with the DO vertically decreased with depths. Overall, the richness tracked variations of DO in linear model and turned at DO concentration of 5.3 mg/L, potentially explaining their turning on the boundary zone of ML and OMZ. The unimodal pattern of bacterial richness with maximum values on the boundary zone of ML and OMZ has been previously reported in the ocean's largest oxygen minimum zone (Beman and Carolan, 2013) and in the seasonal hypoxic Hood Canal (Spietz et al., 2015). This could be explained by the mechanism that as oxygen is depleted, trophic energy is transferred from macrofauna to microbes (Baird et al., 2004; Wright et al., 2012), which allow bacterial taxa that specialized in the hypoxic zone thrive, leading to stimulated richness. Furthermore, the increasing bioavailable energy absorbed to the undegraded organic matter sinking to seafloor allow for existence of multiple taxa resulting in greater biodiversity. The observed correlation between DO and richness implied that microbial aerobic/anaerobic respiration might play an important role in structuring bacterial richness in the open sea.

|
| Fig.7 Correlations between DO and Chao1 richness (a) and Shannon diversity (b) indices (n=66) The blue dotted line is the dividing line of DO more or less than 5.3 mg/L (a) and 4.5 mg/L (b). |
Overall, the community diversity (Shannon diversity indices) tracked variations of DO in polynomial model and peaked at DO concentration of 4.5 mg/L (Fig. 7b), potentially explaining their peaks in the UO. More specifically, when the DO was higher than 4.5 mg/L, diversity increased obviously with the declining DO with depths. In contrast, when the DO was lower than 4.5 mg/L, diversity decreased significantly with the declining DO with depths. Our data, together with previous findings (Beman and Carolan, 2013), suggested that the community diversity of bacterial communities was the greatest in the upper portions of the OMZ, i.e., UO. This was possibly due to the complicated biogeochemical conditions observed there, where both aerobic and anaerobic nitrogen and/or sulfur transformation processes occurred at certain oxygen levels in the OMZ waters. In other words, the boundary region between ML and OMZ, especially the UO, was likely a transition zone where all the aerobic, anaerobic, and microaerophilic communities coexist and perhaps interact. The more diverse bacterial communities in the OMZs than in ML was likely driven by the suboxic and anoxic conditions in the OMZ, which may allow more niche availability. That was to say, microbes can use multiple electron acceptors for respiration in the OMZ waters, such as the Nitrososphaeria were demonstrated a capacity for bacterial growth using ammonia oxidation as an energy source (Zhong et al., 2020). In contrast, in the ML, oxygen is preferentially used and not limiting, reflecting a decrease in niche availability (Pajares et al., 2020).
In conclusion, the bacterial richness possibly increased as the undegraded organic matter sinking to the seafloor, while co-occurrence of aerobic and anaerobic processes in boundary zones of the OMZ within the water column may explain the high diversity at these zones.
4.2 Bacterial community composition altered by DOThe contrasting responding patterns of bacterial alpha-diversity to low and high DO concentrations (Fig. 7) suggested distinct bacterial community composition that predominated the OMZ and other layer, which were accompanied with divergent ecological functions. In the ML, the prevalent Alphaproteobacteria, Cyanobacteria, and Euryarchaeota are known to be responsible for phototrophy, photoautotrophy, and oxygenic photoautotrophy via using proteorhodopsin (Zhang et al., 2019), which corresponded well with our predicted function profiling results (Fig. 6). In fact, the prevalent photoautotrophs in the ML might be result of OMZ impinging on the photic zone and the oxygenic photoautotrophs could provide local oxygen source to feed aerobic processes in anoxic environment, as adaptation to low-oxygen tensions. In the UO and OM, the predominant ammonia-oxidizing Thaumarchaeota (Fig. 4) was consistent with previously reported enrichment of Thaumarchaeota in the hypoxia zone (Gillies et al., 2015; Muck et al., 2019). The highly detected thaumarchaeon Nitrososphaeria suggested the process of ammonium being aerobic oxidated to nitrite in the OMZ and thus was considered to contribute substantially to nitrogen cycling. This was reinforced by the abundant archaeal amoA gene in the OM reported at the same study site (Zhong et al., 2020). Our predicted function profiling result showed that the distribution of Thaumarchaeota corresponded well with that of bacterial OTUs indicating nitrate reduction, nitrate respiration, nitrogen respiration, nitrite respiration and denitrification (Figs. 4 & 6), further supported their contribution to nitrogen cycling in the OMZ water. The sulfate-reducing Deltaproteobacteria have been known to be anaerobic and related to the dark oxidation of sulfur compounds and dark sulfide oxidation (Aldunate et al., 2018), which was reinforced by the predicted function profiling (Fig. 6). Therefore, the Deltaproteobacteria were considered to contribute substantially to sulfur transformation in the OMZ water. In addition, the ubiquitous Gammaproteobacteria throughout the water column were possibly due to their involvement in the (aerobic) chemoheterotrophy.
When the obtained data were pooled together (n=66), bacterial classes do not decrease or increase monotonically with DO but instead fit divergent responding patterns (Fig. 8). For example, the relative abundances of unidentified_Cyanobacteria (family Synechococcales) increased exponentially with the increasing DO and thrived solely at layers where DO>6 mg/L (Fig. 8a), suggesting their strict aerobic metabolism. Alphaproteobacteria showed significant and positive correlation with DO when the DO < 6 mg/L, while remained high abundances when the DO>6 mg/L (Fig. 8b), which suggested that the subgroups are both aerobe and facultative anaerobe. This was consistent with their mixotrophic lifestyle reported in previous finding (Kang et al., 2011), showing that Alphaproteobacteria harbored the metabolic potential for aerobic anoxygenic phototrophy, sulfite-oxidizing chemolithotrophy, CO2 fixation, and CO oxidation. Although no obvious trend to follow with DO variations, Gammaproteobacteria prevalently distributed across the DO gradient between 3 and 5 mg/L and when DO>6 mg/L (Fig. 8c), which suggested that they are both aerobes and anaerobes (Rissanen et al., 2018). Similar pattern has been observed with Gammaproteobacteria throughout the profile, i.e., at order, family and genus levels (Supplementary Fig. S6). In addition, the group has a recognized anaerobic metabolic versatility and the potential for aerobic respiration together with their capacity for use of different carbon sources (Walsh et al., 2009; Aldunate et al., 2018). Moreover, a gamma-proteobacterial sulfur-oxidizing clade (GSO) as the most abundant group within the OMZ core have also been detected by previous work (Stevens and Ulloa, 2008). These findings provided evidence to support versatile metabolisms of these groups and may explain their ubiquity within the water column (Fig. 4). The classes Nitrososphaeria and Deltaproteobacteria presented similar pattern, i.e., they showed polynomial function with variations of DO when DO was between 3 and 5 mg/L (Fig. 8d-e), while remained at low levels when the DO was higher than 6 mg/L. This suggested that these two subgroups were likely anaerobic and facultatively aerobic metabolism within the water column. The kind of niche partition had led to the diversification within these groups, which is the adaptation of these ecotypes that potentially provide a competitive advantage at low-oxygen zone. Moreover, our predictive functional profiling of the bacterial communities highlighted the involvement of these abundant groups in nitrogen and sulfur transformation in the OMZs (Fig. 6, especially in the UO), in line with majority of current research findings (Fernandes et al., 2020). This could be partially supported by the accumulation of nitrite (Supplementary Fig.S5), as previously reported in the anoxic marine zones (Ulloa et al., 2012). Both Acidimicrobiia and Thermoplasmata (Fig. 8f–g) increased gradually with the increasing DO when it was between 3 and 5 mg/L, while showed negligible variation with DO with various abundances when it was higher than 6 mg/L, suggesting that they are aerobic or facultatively anaerobic microorganisms (Golyshina et al., 2016). The classes unidentified_ Actinobacteria, Bacilli and Clostridia had several high values at low DO between 3 and 4 mg/L (Fig. 8h), potentially suggesting their facultatively anaerobic metabolism (Desta et al., 2014).

|
| Fig.8 Correlations between DO and the abundant bacterial classes (n=66) |
These data indicated divergent bacterial assemblages dominate and perform their functional processes of the OMZ and other layers. On the other hand, the presence of aerobic and facultatively anaerobic microorganisms in the OMZ layers, such as Alphaproteobacteria and Gammaproteobacteria (Kang et al., 2011; Aldunate et al., 2018), might be partially explained by their attachment to sinking organic particles. Therefore, the ecological roles of the dominant microbes in the OMZ waters warrant further investigation in future.
4.3 The DO threshold for shift in bacterial communitiesAll the results above-mentioned indicated that the variability of bacterial communities was intimately linked to DO, in consistent with previous findings (Beman and Carolan, 2013; Spietz et al., 2015). Although bacterial communities started to change since the DCM (5.11 to 5.64 mg/L) in parallel with DO, the bacterial community composition actually shifted at the DO concentration of 4.16–4.88 mg/L at 200 m (from being Alphaproteobacteria-dominated to being Gammaproteobacteria-dominated, Fig. 4). The bacterial community threshold for DO (around 4.5 mg/L) were thus identified to be the proposed DO threshold of OMZ in the present study, which is higher than the current standard of hypoxia (2–4 mg/L) (Thomas et al., 2019). Such high DO threshold (5.18– 7.12 mg/L) for shifts in bacterial community structure has been reported in a seasonally hypoxic estuary (Spietz et al., 2015). These finding, together with previous studies, suggested that the shifts in bacterial diversity and community composition, as well their ecological function, preceded the hypoxia effect on the ecological functions of macrofauna. Given the high sensitivity of microbial communities to environmental changes, great cautions should be paid when extrapolating the findings in this study to other biogeochemical processes. In addition, the bacterial taxa such as Nitrososphaeria and Deltaproteobacteria could potentially serve as bioindicator indicating low oxygen area. Due to the limited sampling depths of the stations in the seamount area of TWPO, only ML, UO, and OM were observed for our dataset, which is the drawback of our study. Therefore, further study investigating bacterial properties of the five layers (ML, UO, OM, LO, and SO) in the OMZs is warranted.
5 CONCLUSIONIn the water column of TWPO where OMZs core was clearly identified (with threshold of 3.5 mg/L), bacterial communities tracked the changes of DO and shifted at higher DO concentration (4.5 mg/L) than normal standard (2–4 mg/L). Across stations, bacterial richness and diversity showed unimodal pattern with decreasing DO with depths and peaked at the edge of OMZ. The OMZ harbored contrasting bacterial community composition (the abundant Nitrososphaeria and Deltaproteobacteria) from other layers (the predominant Alphaproteobacteria and Gammaproteobacteria in ML or the dominant Clostridia and unidentified_Actinobacteria in the bottom layer). Through fitting abundant classes with divergent functions with DO, our results demonstrated the sensitivity of key bacterial groups to deoxygenation. Furthermore, the predictive functional profiling highlighted the involvement of abundant groups in nitrogen and sulfur transformation.
The OMZs are globally expanding and intensifying under future climate change, inevitably posing profound effect on pelagic ecosystems. This research study for the first time provided insights into the DOassociated distribution of bacterial assemblages in the TWPO, using high throughput sequence-data. Our findings might have implications for the biogeochemical processes in expanding marine OMZs, which deserves further investigation.
6 DATA AVAILABILITY STATEMENTThe datasets originated from this study are available from the corresponding author with reasonable request.
7 ACKNOWLEDGMENTThe authors thank the crews in R/V Kexue for their supports in data collection and help in the cruise.
Electronic supplementary materialSupplementary material (Supplementary Figs.S1–S6) is available in the online version of this article at https://doi.org/10.1007/s00343-021-0370-0.
Aldunate M, De La Iglesia R, Bertagnolli A D, Ulloa O. 2018. Oxygen modulates bacterial community composition in the coastal upwelling waters off central Chile. Deep Sea Research Part Ⅱ: Topical Studies in Oceanography, 156: 68-79.
DOI:10.1016/j.dsr2.2018.02.001 |
Azam F, Malfatti F. 2007. Microbial structuring of marine ecosystems. Nature Reviews Microbiology, 5(10): 782-791.
DOI:10.1038/nrmicro1747 |
Baird D, Christian R R, Peterson C H, Johnson G A. 2004. Consequences of hypoxia on estuarine ecosystem function: energy diversion from consumers to microbes. Ecological Applications, 14(3): 805-822.
DOI:10.1890/02-5094 |
Beman J M, Carolan M T. 2013. Deoxygenation alters bacterial diversity and community composition in the ocean's largest oxygen minimum zone. Nature Communications, 4: 2705.
DOI:10.1038/ncomms3705 |
Bryant J A, Stewart F J, Eppley J M, DeLong E F. 2012. Microbial community phylogenetic and trait diversity declines with depth in a marine oxygen minimum zone. Ecology, 93(7): 1 659-1 673.
DOI:10.1890/11-1204.1 |
Caporaso J G, Lauber C L, Walters W A, Berg-Lyons D, Lozupone C A, Turnbaugh P J, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the United States of America, 108(S1): 4 516-4 522.
DOI:10.1073/pnas.1000080107 |
Carolan M T, Smith J M, Beman J M. 2015. Transcriptomic evidence for microbial sulfur cycling in the eastern tropical north Pacific oxygen minimum zone. Frontiers in Microbiology, 6: 334.
DOI:10.3389/fmicb.2015.00334 |
Dalsgaard T, Thamdrup B, Farías L, Revsbech N P. 2012. Anammox and denitrification in the oxygen minimum zone of the eastern south Pacific. Limnology and Oceanography, 57(5): 1 331-1 346.
DOI:10.4319/lo.2012.57.5.1331 |
Desta A F, Assefa F, Leta S, Stomeo F, Wamalwa M, Njahira M, Appolinaire D. 2014. Microbial community structure and diversity in an integrated system of anaerobic-aerobic reactors and a constructed wetland for the treatment of tannery wastewater in Modjo, Ethiopia. PLoS One, 9(12): e115576.
DOI:10.1371/journal.pone.0115576 |
Diaz R J, Rosenberg R. 2008. Spreading dead zones and consequences for marine ecosystems. Science, 321(5891): 926-929.
DOI:10.1126/science.1156401 |
Edgar R C. 2013. Uparse: highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10(10): 996-998.
DOI:10.1038/NMETH.2604 |
Fernandes G L, Shenoy B D, Damare S R. 2020. Diversity of bacterial community in the oxygen minimum zones of Arabian Sea and Bay of Bengal as deduced by Illumina sequencing. Frontiers in Microbiology, 10: 3 153.
DOI:10.3389/fmicb.2019.03153 |
Gillies L E, Thrash J C, deRada S, Rabalais N N, Mason O U. 2015. Archaeal enrichment in the hypoxic zone in the northern Gulf of Mexico. Environmental Microbiology, 17(10): 3 847-3 856.
DOI:10.1111/1462-2920.12853 |
Golyshina O V, Kublanov I V, Tran H, Korzhenkov A A, Lünsdorf H, Nechitaylo T Y, Gavrilov S N, Toshchakov S V, Golyshin P N. 2016. Biology of archaea from a novel family Cuniculiplasmataceae (Thermoplasmata) ubiquitous in hyperacidic environments. Scientific Reports, 6: 39 034.
DOI:10.1038/srep39034 |
Gonsalves M J, Paropkari AL, Fernandes C E G, Bharathi PA L, Krishnakumari L, Fernando V, Nampoothiri G E. 2011. Predominance of anaerobic bacterial community over aerobic community contribute to intensify 'oxygen minimum zone' in the eastern Arabian Sea. Continental Shelf Research, 31(11): 1 224-1 235.
DOI:10.1016/j.csr.2011.04.011 |
Hawley AK, Brewer H M, Norbeck AD, Paša-Tolić L, Hallam S J. 2014. Metaproteomics reveals differential modes of metabolic coupling among ubiquitous oxygen minimum zone microbes. Proceedings of the National Academy of Sciences of the United States of America, 111(31): 11 395-11 400.
DOI:10.1073/pnas.1322132111 |
Hoerling M P, Hurrell J W, Xu T Y. 2001. Tropical origins for recent north Atlantic climate change. Science, 292(5514): 90-92.
DOI:10.1126/science.1058582 |
Jain A, Bandekar M, Gomes J, Shenoy D, Meena R M, Naik H, Khandeparkar R, Ramaiah N. 2014. Temporally invariable bacterial community structure in the Arabian Sea oxygen minimum zone. Aquatic Microbial Ecology, 73(1): 51-67.
DOI:10.3354/ame01704 |
Jayakumar A, O'Mullan G D, Naqvi S W A, Ward B B. 2009. Denitrifying bacterial community composition changes associated with stages of denitrification in oxygen minimum zones. Microbial Ecology, 58(2): 350-362.
DOI:10.1007/s00248-009-9487-y |
Kang I, Vergin K L, Oh H M, Choi A, Giovannoni S J, Cho J C. 2011. Genome sequence of strain HTCC2083, a novel member of the marine clade Roseobacter. Journal of Bacteriology, 193(1): 319-320.
DOI:10.1128/jb.01268-10 |
Li X G, Song J M, Yuan H M, Li N, Duan L Q, Wang Q D. 2017. The oxygen minimum zones (OMZs) and its ecoenvironmental effects in ocean. Marine Sciences, 41(12): 127-138.
(in Chinese with English abstract) DOI:10.11759/hykx20170821003 |
Ma J, Song J M, Li X G, Yuan H M, Li N, Duan L Q, Wang Q D. 2019. Environmental characteristics in three seamount areas of the tropical western Pacific Ocean: focusing on nutrients. Marine Pollution Bulletin, 143: 163-174.
DOI:10.1016/j.marpolbul.2019.04.045 |
Ma J, Song J M, Li X G, Yuan H M, Li N, Duan L Q, Wang Q D. 2020. Control factors of DIC in the Y3 seamount waters of the Western Pacific Ocean. Journal of Oceanology and Limnology, 38(C7): 1 215-1 224.
DOI:10.1007/s00343-020-9314-3 |
Muck S, De Corte D, Clifford E L, Bayer B, Herndl G J, Sintes E. 2019. Niche differentiation of aerobic and anaerobic ammonia oxidizers in a high latitude deep oxygen minimum zone. Frontiers in Microbiology, 10: 2 141.
DOI:10.3389/fmicb.2019.02141 |
Naqvi S W A. 1994. Denitrification processes in the Arabian Sea. Proceedings of the Indian Academy of Sciences-Earth and Planetary Sciences, 103(2): 279-300.
DOI:10.1007/BF02839539 |
Pajares S, Varona-Cordero F, Hernández-Becerril D U. 2020. Spatial distribution patterns of bacterioplankton in the oxygen minimum zone of the tropical Mexican Pacific. Microbial Ecology, 80(3): 519-536.
DOI:10.1007/s00248-020-01508-7 |
Paulmier A, Ruiz-Pino D, Garçon V. 2011. CO2 maximum in the oxygen minimum zone (OMZ). Biogeosciences, 8(2): 239-252.
DOI:10.5194/bg-8-239-2011 |
Paulmier A, Ruiz-Pino D. 2009. Oxygen minimum zones (OMZs) in the modem ocean. Progress in Oceanography, 80(3-4): 113-128.
DOI:10.1016/j.pocean.2008.08.001 |
R Core Team. 2013. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria.
|
Rissanen A J, Saarenheimo J, Tiirola M, Peura S, Aalto S L, Karvinen A, Nykänen H. 2018. Gammaproteobacterial methanotrophs dominate methanotrophy in aerobic and anaerobic layers of boreal lake waters. Aquatic Microbial Ecology, 81(3): 257-276.
DOI:10.3354/ame01874 |
Spietz R L, Williams C M, Rocap G, Horner-Devine M C. 2015. A dissolved oxygen threshold for shifts in bacterial community structure in a seasonally hypoxic estuary. PLoS One, 10(8): e0135731.
DOI:10.1371/journal.pone.0135731 |
Stevens H, Ulloa O. 2008. Bacterial diversity in the oxygen minimum zone of the eastern tropical south Pacific. Environmental Microbiology, 10(5): 1 244-1 259.
DOI:10.1111/j.1462-2920.2007.01539.x |
Sun Q Q, Song J M, Li X G, Yuan H M, Ma J, Wang Q D. 2020. Bacterial vertical and horizontal variability around a deep seamount in the tropical western Pacific Ocean. Marine Pollution Bulletin, 158: 111 419.
DOI:10.1016/j.marpolbul.2020.111419 |
Thomas Y, Flye-Sainte-Marie J, Chabot D, Aguirre-Velarde A, Marques G, Pecquerie L. 2019. Effects of hypoxia on metabolic functions in marine organisms: observed patterns and modelling assumptions within the context of dynamic energy budget (DEB) theory. Journal of Sea Research, 143: 231-242.
DOI:10.1016/j.seares.2018.05.001 |
Tian D F, Li X G, Song J M, Li N. 2019. Process and mechanism of nitrogen loss in the ocean oxygen minimum zone. Chinese Journal of Applied Ecology, 30(3): 1 047-1 056.
(in Chinese with English abstract) DOI:10.13287/j.1001-9332.201903.038 |
Tian D F, Wang Y Q, Xing J W, Sun Q Q, Song J M, Li X G. 2020. Nitrogen loss process in hypoxic seawater based on the culture experiment. Marine Pollution Bulletin, 152: 110 912.
DOI:10.1016/j.marpolbul.2020.110912 |
Ulloa O, Canfield D E, Delong E F, Letelier R M, Stewart F J. 2012. Microbial oceanography of anoxic oxygen minimum zones. Proceedings of the National Academy of Sciences of the United States of America, 109(40): 15 996-16 003.
DOI:10.1073/pnas.1205009109 |
Ulloa O, Wright J J, Belmar L, Hallam S J. 2013. Pelagic oxygen minimum zone microbial communities. In: Rosenberg E, DeLong E F, Lory S, Stackebrandt E, Thompson F eds. The Prokaryotes. Springer, Berlin, Heidelberg, p. 113-122, https://doi.org/10.1007/978-3-642-30123-0_45.
|
Walsh D A, Zaikova E, Howes C G, Song Y C, Wright J J, Tringe S G, Tortell P D, Hallam S J. 2009. Metagenome of a versatile chemolithoautotroph from expanding oceanic dead zones. Science, 326(5952): 578-582.
DOI:10.1126/science.1175309 |
Wang Q, Garrity G M, Tiedje J M, Cole J R. 2007. Naïve Bayesian classifier for rapid assignment of RRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73(16): 5 261-5 267.
DOI:10.1128/AEM.00062-07 |
Wishner K F, Outram D M, Seibel B A, Daly K L, Williams R L. 2013. Zooplankton in the eastern tropical north Pacific: boundary effects of oxygen minimum zone expansion. Deep Sea Research Part I: Oceanographic Research Papers, 79: 122-140.
DOI:10.1016/j.dsr.2013.05.012 |
Wright J J, Konwar K M, Hallam S J. 2012. Microbial ecology of expanding oxygen minimum zones. Nature Reviews Microbiology, 10(6): 381-394.
DOI:10.1038/nrmicro2778 |
Zhang W, Liu J, Dong Y, et al. 2019. Archaeal community structure in sediments from a seamount in the Mariana volcanic arc. Journal of Oceanology and Limnology, 37(4): 1 197-1 210.
DOI:10.1007/s00343-019-8044-x |
Zhong H H, Lehtovirta-Morley L, Liu J W, Zheng Y F, Lin H Y, Song D L, Todd J D, Tian J W, Zhang X H. 2020. Novel insights into the Thaumarchaeota in the deepest oceans: Their metabolism and potential adaptation mechanisms. Microbiome, 8(1): 78.
DOI:10.1186/s40168-020-00849-2 |
Zuo J L, Song J M, Yuan H M, Li X G, Duan L Q. 2019. Impact of Kuroshio on the dissolved oxygen in the East China Sea region. Journal of Oceanology and Limnology, 37(2): 513-524.
DOI:10.1007/s00343-019-7389-5 |