 2021, Vol. 39
2021, Vol. 39Institute of Oceanology, Chinese Academy of Sciences
Article Information
- HE Yuan, SHEN Songdong, YU Dachun, WANG Yehua, YIN Jiao, WANG Zongling, YE Yuantu
- The Ulva prolifera genome reveals the mechanism of green tides
- Journal of Oceanology and Limnology, 39(4): 1458-1470
- http://dx.doi.org/10.1007/s00343-020-0212-5
Article History
- Received May. 29, 2020
- accepted in principle Jul. 22, 2020
- accepted for publication Sep. 23, 2020




2 First Institute of Oceanography(FIO), Ministry of Natural Resources(MNR), Qingdao 266100, China
Chlorophyta is an important group of green algae, with approximately 350 genera and 5 000–8 000 species. The green algae group can be divided into two classes: Chlorophyceae and Conjugatophyceae. Green algae present a variety of forms, including single-cell individuals, filaments, and membranous bodies (Mine et al., 2008). Most green algae live in fresh water, whereas only approximately 10% grow in seawater. In China, seawater green algae belong to Chlorophyceae, which is divided into 11 orders, namely, Volvocales, Tetrasporales, Ulotrichales, Chlorococcales, Ulvales, Chaetophorales, Cladophorales, Siphonales, Siphonocladales, Prasiolales, and Dasycladales. The cell wall of green algae contains cellulose, and the cytological features share many similarities with those of higher vascular plants. Furthermore, the pigments produced are the same photosynthetic pigments of higher plants, such as chlorophyll a, chlorophyll b, carotene, and lutein. The chloroplasts of green algae contain pyrenoids, which are surrounded by starch. Previous evolutionary analyses show that Chlorophyta is more closely related to higher plants than other algae, and higher plants are likely to have evolved from green algae (Herron et al., 2010).
Ulva mainly grows on rocks in intertidal zone on mud beaches or onto other floating seaweeds at the sea (Tan et al., 1999). There are approximately 11 species reported in China. The common species are U. pertusa, U. lactuca, U. prolifera, U. intestinalis, U. compressa, U. tubulosa, U. flexuosa, and U. linza. U. prolifera belongs to phylum Chlorophyta, class Ulvophyceae, order Ulvales, family Ulvaceae, and genus Ulva (Gao et al., 2010). Algal bodies are bright green or light green, in height of 1–2 m and diameters 2–3 mm, with many branches. The main branches are distinct and elongated and have typical tubular structure of monolayer cells. The pigment body is not filled with cells and contains one starch core.
In life history, U. prolifera exhibits a variety of reproductive manners, such as asexual reproduction (vegetative reproduction and spore reproduction), sexual reproduction, and parthenogenesis.
Regarding biological characteristics, U. prolifera could tolerate wide ranges of temperature and salinity, and resist to strong light. Previous studies have shown that the adaptability of U. prolifera to the environment is within the following conditions: temperature 10– 30 ℃, salinity 7.2–53.5, light 1 000–10 000 lx, and pH 6–9 (Gao et al., 2016). Reports of U. prolifera cultured in China indicate the ash content of 8.2%– 36.68%, which is lower than that of kelp and wakame and higher than that of seaweed, and the vitamin content of 0.174–0.206 mg/g, which is higher than that of kelp and wakame. Moreover, U. prolifera is rich in protein and has high digestibility (98%). Therefore, it can be used as an important protein food source for humans. U. prolifera also contains 18 amino acids, including all 8 essential amino acids (Xu et al., 2003). Sugars account for the largest proportion of U. prolifera cells, at 46.2%–63.9%, and become polymerized as dietary fiber in a certain structure that is a good source of dietary fiber. Moreover, as this species synthesizes a wide range of fatty acids, including linolenic acid, linoleic acid, eicosapentaenoic acid (EPA), arachidonic acid, and other important fatty acids, it can be used as a raw material for producing functional fats and oils. In addition, levels of three important mineral elements, Fe, Zn, and Cu, are significantly higher in U. prolifera than in other algae. Therefore, this species can be used as a good feed source (Fleurence et al., 1994).
Algae comprise an ancient group. These species have crossed the prokaryotic and eukaryotic boundary. To date, there are approximately 30 000 known species. Indeed, the evolutionary relationship of algae has been a heavily researched topic for many years (Ben Ali et al., 2002). With the development of genome sequencing technology and the decreasing cost, there have been some recent breakthroughs in genomics research of algae, and the whole genomes of several algae have been published.
In this study, the whole-genome sequence of U. prolifera was studied with a focus on genes that involved in growth and stress resistance. In addition, our data were compared with those of U. mutabilis, which is available in GenBank. The findings could not only enrich our knowledge of Ulva genomes but also provide a foundation for understading the U. prolifera outbreaks in terms of the cell division process.
2 MATERIAL AND METHOD 2.1 Strain and culture conditionsFloating green microalga U. prolifera was collected in June 2018 at the Yellow Sea in Qingdao, Shandong, China (34°5ʹN, 122°50ʹE). The sample was placed on dry ice, transported to laboratory, and cultivated in seawater medium. The cultivation environment was 20 ℃, salinity 30, under cool-white fluorescent light in 12 h꞉12 h L꞉D cycle, illuminance 120 μmol/(m2·s) photons, and the length of culture 7 d. After the culture, they were frozen in liquid nitrogen for DNA extraction.
2.2 Genome sequencing and assemblyTotal DNA was isolated using E.Z.N.A.® Plant DNA Kit (Omega BioTek, USA) as per the manufacturer's protocol. A 20-kb DNA library was prepared according to PacBio DNA preparation protocols, and the whole genome was sequenced using the PacBio RS platform. De novo assembly of the genome was achieved using falcon software (Chin et al., 2016). The original reads were split into modules of specified sizes and compared. Line selfcorrection was employed, and overlapping data were searched. The Overlap-Layout-Consensus algorithm was applied to assemble the three generations of data after error correction through the overlap relationship among long reads. The original reads to the preliminary assembly results was mapped by correcting the latter to improve the accuracy. The assembly and annotation data were uploaded to NCBI under the accession number SDUY00000000.1.
2.3 Genome annotation and analysisGenome annotation mainly includes three aspects: noncoding RNA prediction, gene structure prediction, and gene function annotation.
Noncoding RNAs include sRNA, rRNA, tRNA, snRNA, and miRNA. tRNAs were predicted using tRNAscan-SE (v1.3.1) (Schattner et al., 2005) and rRNAs using RNAmmer (v1.2) (Lagesen et al., 2007). Gene structure prediction of the genome was performed after masking repetitive sequences using the de novo prediction method with AUGUSTUS (Stanke et al., 2004). The reference genome used for the annotation was that of U. mutabilis (De Clerck et al., 2018). Gene function annotation was carried out using the NR, COG/KOG, GO, KEGG, Swiss-Prot, and eggNOG databases. Alignment was performed using diamond software (Buchfink et al., 2015), with e < 1e-5 annotations; proteins with the highest sequence similarity were screened to obtain functional annotation information.
2.4 Repeated sequence analysisRepeated sequences can be divided into two major types, tandem repeats and interspersed repeats, depending on their arrangement in the genome (Wickstead et al., 2003). Repeated sequences were predicted using RepeatMasker (v4.0.7) software.
2.5 Phylogenetic tree reconstructionThe genomes of some green algae have been published in NCBI public databases. To compare homologous sequences, we first assembled each genome for alignment and then examined homologous sequences. We combined the genome of U. prolifera obtained in this study with other green alga genomes and constructed a phylogenetic tree at the genome level. A maximum likelihood tree was constructed by using the software RAxML.
2.6 Analysis of genes related to growth and stress resistanceGenes involved in the cell cycle were analyzed using KEGG pathway annotation, and those involved in phosphorylation were identified using GO database annotation. Some genes related to stress resistance were found through the NR database.
3 RESULT 3.1 Genome sequencing and annotationWe first generated a total of 2 213 533 reads with a total size of 12 897 726 267 bp, a minimum length of 50 bp, a maximum length of 83 142 bp, and a mean length of 5 901 bp. After removing redundant scaffolds from the initial assembly, the N50 and N75 were 675 063 and 277 886 bp, respectively. The assembly of the U. prolifera nuclear genome was 87.9 Mb, with contained 597 contigs (Table 1). The largest contig was 2 662 757 bp, with a GC content of 55.92%. The integrity of the genomic sequences and genetic predictions were analyzed using BUSCO software (Simão et al., 2015). The integrity of the genome was 75.3%.

Noncoding RNAs identified included rRNA, tRNA, and the other ncRNA. The total number of noncoding RNAs was 557. In addition, 185 rRNAs were found, accounting for 33.21%; 319 tRNAs, accounting for 57.27%; and 53 other ncRNAs, accounting for 9.52%. A total of 10 311 genes were detected, with a total length of 17 932 935 bp on average of 1 739 bp. 10 311 unigenes were annotated using different databases (NR, Swiss-Prot, KEGG, KOG, eggNOG, and GO), with matches of 7 037 (68.25%) in NR, 4 856 (47.10%) in Swiss-Prot, 3 315 (32.15%) in KEGG, 4 495 (43.59%) in KOG, 6 180 (59.94%) in eggNOG, and 4 770 (46.26%) in GO.
A total of 3 315 unigenes were annotated in the KEGG database, involving 24 metabolic pathways (Fig. 1). The pathway with the most unigenes was "translation" (324 unigenes), followed by "global and overview maps" (273 unigenes) and "energy metabolism" (262 unigenes). There were 114 unigenes predicted to participate in "cell growth and death", including "cell cycle", "cell cycle-yeast", "cell cyclecaulobacter", "meiosis-yeast", "oocyte meiosis", "p53 signaling pathway", "apoptosis", "apoptosisfly", and "apoptosis-multiple species". Genes of interested are involved in "cell cycle".

|
| Fig.1 KEGG annotation of the nonredundant sequences of the sample The y-axis indicates the name of the KEGG metabolic pathway. The x-axis indicates the number of genes. The unigenes annotated were involved in 24 metabolic pathways. |
GO functional annotations consist of three ontologies: biological process, cellular component, and molecular function. In total, 4 770 annotated unigenes were categorized into the three ontolog with 64 GO terms (Fig. 2). Most unigenes are involved in the "cellular component" category (4 453 unigenes) followed by "molecular function" (4 202 unigenes) and "biological process" (4 053 unigenes). For the cellular component category, "cell" and "cell part" were two most common terms; "binding" and "catalytic activity" were the two most common terms in the molecular function category and "cellular process" and "metabolic process" in the biological process category.

|
| Fig.2 GO annotation of the nonredundant sequences of the sample Three primary GO categories and 64 subcategories were summarized in the GO database. |
The proportion of repeated sequences in the genome was 21.92% by calculation. Among the sequences, 84 small RNAs are present in tandem repeats, accounting for 0.07% of the total number of sequences. There are 19 009 simple repeats, accounting for 1.23%, and 908 low-complexity sequences, accounting for 0.05%. The total length of the interspersed repeat is 18 100 769 bp, with the largest number of the total number of sequences being long terminal repeats (LTRs), at 4 353, accounting for 6.43%, followed by DNA elements, at 2 653, accounting for 0.78%, and long interspersed nuclear elements (LINEs), at 1 026, accounting for 0.48%. Short interspersed nuclear element sequences (SINEs) represented the least, at 205, accounting for 0.41%, and there were 35 564 unclassified tandem repeats.
3.3 Phylogenetic tree analysisTo better understand the evolutionary status of U. prolifera among green algae, the genomes of other green algae present in GenBank, such as Volvox, Chlamydomonas, Chlorella, Dunaliella, and Gonium, were used to construct phylogenetic trees. Arabidopsis and rice clustered together and acted as an outgroup. Eighteen species of green algae were divided into four categories: Chlorophyta, Ulvophyceae, Trebouxiophyceae, and Prasinophyceae (Fig. 3). The genomes of U. prolifera and U. mutabilis were the closest, clustering together with Ulvophyceae. The result also supports the general notion that Chlorophyceae and Ulvophyceae are evolutionarily close within Chlorophyta.

|
| Fig.3 The phylogenetic tree was constructed based on the genomes of U. prolifera and the other green algae using maximumlikelihood methods |
Ulva is the representative of Ulvophyceae. At present, only one Ulva species, namely, U. mutabilis, has been fully sequenced (De Clerck et al., 2018). To explore the features of the Ulva genome, we compared our data with that of U. mutabilis.
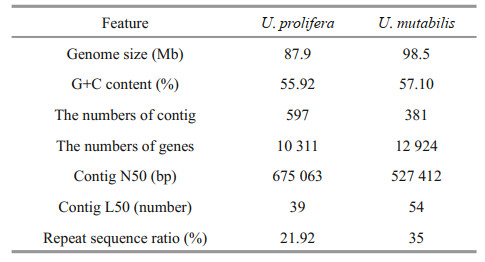
The size of the genome of U. mutabilis is 98.5 Mb, which is more than 10.6 Mb larger than the genome of U. prolifera. The number of protein-coding genes is 12 924, which is also more than that obtained for U. prolifera (10 311). However, the N50 of U. prolifera is 675 063 bp, which is longer than that of U. mutabilis (527 412 bp). In addition, the GC content of U. mutabilis is 57.1%, which is similar to that of U. prolifera (55.92%). Based on the GC content and morphological complexity, the morphological features of the two species are very similar. In terms of repetitive sequences, LTRs and LINEs in U. mutabilis comprise 15.3 Mb and 9.3 Mb, respectively, which is considerably longer than the values for U. prolifera (5.9 Mb and 0.5 Mb). A comparison of the general features of the U. prolifera and U. mutabilis genomes is presented in Table 2. The result of collinear analysis also showed that the genetic type and relative sequences of the two algae are relatively conserved (Fig. 4).

|
| Fig.4 Collinear analysis of U. prolifera and U. mutabilis |
Fifty genes are involved in the cell cycle according to KEGG annotation, and we found 27 cell cycle pathway-related genes in algae via comparison with data in GenBank (Table 3). These genes included some cell cycle factors, such as CDC14, CCNA, CDC2, and CDC23, cyclin-dependent kinase proteins, such as CDKD, other genes involved in the cell cycle, such as MCM2-7, and anaphase-promoting complexes, such as APC1 and APC6.

Based on annotations, a large number of genes were identified as being responsive to stress, as follows: heat shock protein 40 in response to hightemperature stress; a cold shock protein in response to low-temperature stress; phytoene desaturase and carotenoid isomerase in response to high light; and catalase, ascorbate peroxidase, glutathione peroxidase, thioredoxin reductase, dehydroascorbate reductase, peroxiredoxin-5, peroxiredoxin-2B, ironsuperoxide dismutase 2, iron-superoxide dismutase 1, glutathione reductase, and iron reductase related to antioxidants. Some ion channel proteins were also identified, including chloride channel protein, voltage-gated channel, cadmium channel protein, and sodium channel protein.
4 DISCUSSIONThe first species in Rhodophyta with a sequenced genome was Cyanidioschyzon merolae, and the results indicate that the C. merolae genome is an ideal model for studying the origin, evolution, and basic mechanisms of eukaryotes (Matsuzaki et al., 2004). Genome-sequencing research of Pyropia yezoensis has also been completed, with a size of 43 Mb. In total, 10 327 genes were predicted, similar to the number of genes in U. prolifera in the present results. Sequence homology analysis indicated an unknown function for 3 611 genes, of which 2 069 genes were identical to those of C. merolae, and the light-trapping pigment phycobilin was predicted determinant of the pigment characteristics of red algae. Thus, the genome of P. yezoensis is an ideal model of Rhodophyta (Nakamura et al., 2013). As shown in our data, the genome of U. prolifera is larger than that of P. yezoensis. The genome of P. umbilicalis is 87.7 Mb, similar to that of U. prolifera including 13 125 genetic loci, although this number is greater than that of U. prolifera. The findings provide an important reference for the comprehensive understanding of the evolutionary relationship of red algae with other algae associated with secondary endosymbiosis (Brawley et al., 2017). The genome of Chondrus crispus has also been sequenced, with a size of 105 Mb and 9 606 annotated genes. Although the Chondrus crispus genome size is larger than that of U. prolifera, the number of genes is slightly smaller than that in U. prolifera. These genomic data provide insight into the metabolic characteristics of marine red algae and their ability to adapt to the marine environment, including halogen metabolism, hydroxyl phospholipids, and multicellularity (Collén et al., 2013).
The genome of Ectocarpus siliculosus was the first sequenced in Phaeophyta, and its size was 214 Mb reportedly, much larger than that of U. prolifera. The discovery of light-harvesting and pigment-synthesizing genes and new metabolic processes, including halide metabolism, can help explain why E. siliculosus is able to adapt to the high degree of change in tidal environment. Overall, previous studies indicate that E. siliculosus is an ideal model for studying the genome of brown algae (Cock et al., 2010). In the future, we will explore whether similar metabolic processes exist in U. prolifera to adapt to various sea environment. The complete kelp genome in size of 537 Mb and 18 733 annotated genes, has also recently been obtained. The size of the genome and number of genes are much larger than that in U. prolifera. This research made important contributions to plant genome research (Ye et al., 2015). Ectocarpus confervoides has a genome size of 140 Mb, containing 13 640 protein-coding genes; this genome is smaller than those of E. siliculosus and kelp but larger than that of U. prolifera (Nishitsuji et al., 2016).
With a size of 34 Mb, the genome of Thalassiosira pseudonana was the first to be sequenced in Bacillariophyta, and 24 chromosomes were obtained by sequence assembly (Mock et al., 2008). Phaeodactylum tricornutum, with a genome size of 27.4 Mb, is a model organism in Bacillariophyta, and it was found significantly different from the genomic structure of T. pseudonana (Bowler et al., 2008). The genome sizes of algae in Bacillariophyta are generally smaller than that of U. prolifera, and the structures are relatively simple.
In Chlorophyta, the genomes of many species have been successfully sequenced. A model organism of green algae, the genome size of Chlamydomonas reinhardtii is 120 Mb. In general, studying the genome can deepen our understanding of primitive eukaryotic cells and reveal previously unknown genes associated with photosynthesis and the function of flagella; indeed, a link between ciliary diseases and the components and functions of the flagella has been established, showing the characteristics of the last common ancestor of plants and animals and identifying many cilia- and plastid-related genes (Merchant et al., 2007). Moreover, the genomes of three species of Chlorella have been determined. Chlorella variabilis NC64A, with a genome size of 46 Mb, is an ideal model for studying the interaction between algae and viruses. The symbiotic protein family was found to have greatly improved our understanding of the evolution of green algae, including interactions with viral genomes and symbiotic relationships with other eukaryotes (Blanc et al., 2010). Chlorella protothecoides sp. 0710 is considered one of the best candidates for industry production of microalgal biofuel. Its genome size is 22.9 Mb, which is only half of that of C. variabilis. Genomic studies of C. protothecoides have revealed the mechanism underlying its capacity to produce large amounts of lipids, and genome analysis has facilitated evaluating algae as a model for the production of oils. With a genome size of 56.8 Mb, C. pyrenoidosa FACHB-9 is a microalga that rapidly converts intracellular stored energy to lipids. These studies may provide a basis for investigating metabolism and optimizing food and fuel production (Fan et al., 2016). The genome size of Monoraphidium neglectum is 68 Mb. Its carbohydrate metabolism and fatty acid biosynthesis processes, which are used in biotechnology for carbohydrate conversion, exhibit high metabolic diversity and provide new insight into the metabolic diversity of microalgal lipids (Bogen et al., 2013). Additionally, Parachlorella kessleri is a high-efficiency unicellular green alga in genome size of 62.5 Mb. Upregulation of synthesis and autophagy is a potential key mediator of lipid hyperaccumulation under nutrient stress conditions (Ota et al., 2016). Volvox carteri is a multicellular green alga that is suitable for studying the evolution and development of multicellular organisms; its genome size is 138 Mb. There are only a small number of specific protein-coding genes in V. carteri compared with C. reinhardtii; the C. reinhardtii genome encodes some proteins involved in the degree of expansion and height during division as well as the extracellular matrix. In general, the increased complexity of organisms is associated with modification of pedigree-specific proteins, rather than a large increase in protein-coding ability (Prochnik et al., 2010). Gonium pectorale is another species of Volvocales, with a genome size of 148.8 Mb, similar to that of Volvox carteri. The evolution of cell cycle regulation is important for population evolution and may reveal the evolutionary history of other multicellular evolutionary transitions (Hanschen et al., 2016). Ostreococcus tauri and Ostreococcus lucimarinus are two small eukaryotic green algae. O. tauri has a genome size of 12.56 Mb, and some special features and a streamlined gene family make O. tauri an ideal model for studying the evolution of eukaryotic genes, including the origins of specific chromosomes and green algae families (Derelle et al., 2006). At 13.2 Mb, the genome size of O. lucimarinus is similar to that of O. tauri, and the study of the O. lucimarinus genome has offered insight into the biospecific metal metabolism of these organisms, which contain large amounts of selenocysteine proteins (Palenik et al., 2007). Micromonas is a genus of very small green algae with diameters of less than 2 microns. The genomes of Micromonas RCC299 and Micromonas CCMP1545 were sequenced simultaneously, with sizes of 20.9 Mb and 21.9 Mb, respectively, which are considerably smaller than that of C. reinhardtii but larger than that of Ostreococcus. This finding highlights the dynamics of the evolution of marine algae and provides a platform for further exploring the functions of phytoplankton (Worden et al., 2009). Dunaliella salina is a single-celled green alga that is extremely tolerant to high salt. The genome of D. salina CCAP19/18, 343.7 Mb, was recently published, and the study of this genome facilitated the investigation of metabolic processes involved in the regulation of stress responses, including carotenoid production and growth in high-salt environments (Polle et al., 2017). The genomes of Guillardia theta and Bigelowiella natans are 87.2 Mb and 94.7 Mb, respectively (Curtis et al., 2012). Coccomyxa subellipsoidea is a green alga that grows in the polar region and has strong cold adaptability. Its genome size is 48.8 Mb, and this is the first determination of polar eukaryotic microorganisms. Moreover, it was found that prokaryotes and eukaryotes followed a similar evolutionary route to achieve survival under cold conditions (Blanc et al., 2012). The genome size of U. prolifera is moderate among members of Chlorophyta, and it is very close to that of Guillardia theta.
In this study, we sequences the genome of U. prolifera, with a size of 87.9 Mb, and the data obtained were compared with those for U. mutabilis. U. prolifera is the main species that causes green tides (Wu et al., 2018), which have negative impacts on the environment and local economy in China every year. Therefore, the reasons why U. prolifera can cause green tides have been a heavily researched topic in the study of green algae. We identified the genes involved in the cell cycle, which includes the entire process from the completion of one division to the end of the next division and is divided into a division phase and an interphase. Regulation of the cell cycle is an important aspect of biological growth and development, and many important life processes are related to the cell cycle. However, aberrant regulation leads to excessive cell proliferation or apoptosis. Accordingly, cell division is critical to organisms, and protein kinase-mediated signal transduction is the primary means of cell cycle regulation. Cyclin (cdc) and cyclin-dependent kinase (cdk) are key kinases that regulate the cell cycle (Joubès et al., 2000). Cyclins comprise a heterogeneous family of proteins, the main feature of which is the ability to bind and activate members of the cyclin-dependent kinase (CDKs) family and participate in cell cycle regulation (John et al., 2001). We detected some related genes in the genome of U. prolifera, including some cell cycle proteins. In general, the cell division activity of U. prolifera cells is vigorous, and rapid growth may result from high cell proliferation and differentiation activity.
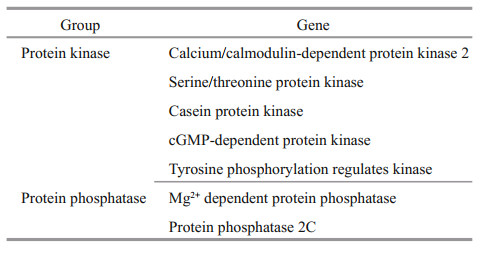
Phosphorylation is the most important posttranslational modification (Yang, 2005; Hunter, 2007; Temporini et al., 2008). Some protein phosphatase and protein kinase genes were identified in this study, and these genes constitute a switch system for phosphorylation and dephosphorylation (Table 4). It has been predicted that approximately 30% of the eukaryotic cell proteome is phosphorylated (Cohen, 1976). In fact, phosphorylation is involved in many key processes, such as intercellular communication, immune response, intracellular signal transduction, and epigenetic regulation, and this process plays an important role in the growth and environmental response of U. prolifera.
The reason why U. prolifera is able to grow rapidly in a short period is closely related to its capacity to adapt to various hostile environments; that is, it possesses strong resistance to stress. Under lowtemperature stress, lipid molecules on the plasma membrane will change to adapt to a low-temperature environment (Chen et al., 2005). Under hightemperature conditions, plants have a heat shock response to maintain homeostasis, protecting cells and normal biological activity and thereby enhancing resistance to heat (Liu and Wang, 2006). When the active oxygen species produced exceeds the ability of the scavenging system, oxidative damage and even cell death ensue (Du et al., 2001; Saxena et al., 2016). Algae, similar to other plants, have evolved protective enzymes that scavenge free radicals and reactive oxygen (Sies, 1993), and antioxidant enzymes coordinate to maintain biological free radicals at low levels to prevent cell damage (Zelko et al., 2002; Zhang and Tian, 2007). The discovery of such stressresponsive genes in U. prolifera indicates that the alga has strong resistance to stress, and even under adverse growth conditions, this species can rely on the expression of these stress-resistant genes to effectively resist external unfavorable factors. This is consistent with the characteristic of strong survivability, supporting the premise that U. prolifera can grow vigorously in nature.
5 CONCLUSIONThe green alga U. prolifera is the main species responsible for green tides in the Yellow Sea. Here, we report an annotated draft genome of U. prolifera, which is a candidate model alga of Ulva. Comparative analysis of our data with that of U. mutabilis revealed a close genetic relationship between the two species; however, some genomic differences exist between them, such as genome size and number of genes. To explore the evolutionary status of U. prolifera, we constructed a phylogenetic tree using the genomes of green algae published in GenBank. Some genes involved in the cell cycle, phosphorylation, and stress resistance were found in U. prolifera, suggesting that its rapid growth is associated with these genes, which may explain the reason for green tides from a new perspective. These findings will help to elucidate the biology of U. prolifera and will serve as a foundation for exploring the evolutionary history of other algae in Ulva.
6 DATA AVAILABILITY STATEMENTSequence data that support the findings of this study have been deposited in the National Center for Biotechnology Information (NCBI) with the accession number SDUY00000000.1.
7 AUTHOR DECLARATIONThe authors declared no conflict of interest. No conflicts, informed consent, human or animal rights applicable was concerned.
All authors have agreed to authorship and the submission of this manuscript for peer review. Yuan HE performed the experiments, analyzed the data, and drafted the manuscript; Songdong SHEN designed the experiments; Dachun YU, Yehua WANG, and Jiao YIN collected the samples; Zongling WANG analyzed the data; and Yuantu YE reviewed the manuscript.
Ben Ali A, De Baere R, De Wachte R, Van de Peer Y. 2002. Evolutionary relationships among heterokont algae (the autotrophic stramenopiles) based on combined analyses of small and large subunit ribosomal RNA. Protist, 153(2): 123-132.
DOI:10.1078/1434-4610-00091 |
Blanc G, Agarkova I, Grimwood J, Kuo A, Brueggeman A, Dunigan D D, Gurnon J, Ladunga I, Lindquist E, Lucas S, Pangilinan J, Pröschold T, Salamov A, Schmutz J, Weeks D, Yamada T, Lomsadze A, Borodovsky M, Claverie J M, Grigoriev I V, Van Etten J L. 2012. The genome of the polar eukaryotic microalga Coccomyxa subellipsoidea reveals traits of cold adaptation. Genome Biology, 13(5): R39.
DOI:10.1186/gb-2012-13-5-r39 |
Blanc G, Duncan G, Agarkova I, Borodovsky M, Gurnon J, Kuo A, Lindquist E, Lucas S, Pangilinan J, Polle J, Salamov A, Terry A, Yamada T, Dunigan D A, Grigoriev I V, Claverie J M, van Etten J L. 2010. The Chlorella variabilis NC64A genome reveals adaptation to photosymbiosis, coevolution with viruses, and cryptic sex. Plant Cell, 22(9): 2943-2955.
DOI:10.1105/tpc.110.076406 |
Bogen C, Al-Dilaimi A, Albersmeier A, Wichmann J, Grundmann M, Rupp O, Lauersen K J, Blifernez-Klassen O, Kalinowski J, Goesmann A, Mussgnug J H, Kruse O. 2013. Reconstruction of the lipid metabolism for the microalga Monoraphidium neglectum from its genome sequence reveals characteristics suitable for biofuel production. BMC Genomics, 14(1): 926.
DOI:10.1186/1471-2164-14-926 |
Bowler C, Allen A E, Badger J H, Grimwood J, Jabbari K, Kuo A, Maheswari U, Martens C, Maumus F, Otillar R P, Rayko E, Salamov A, Vandepoele K, Beszteri B, Gruber A, Heijde M, Katinka M, Mock T, Valentin K, Verret F, Berges J A, Brownlee C, Cadoret J P, Chiovitti A, Choi C J, Coesel S, De Martino A, Detter J C, Durkin C, Falciatore A, Fournet J, Haruta M, Huysman M J J, Jenkins B D, Jiroutova K, Jorgensen R E, Joubert Y, Kaplan A, Kröger N, Kroth P G, La Roche J, Lindquist E, Lommer M, Martin-Jézéquel V, Lopez P J, Lucas S, Mangogna M, McGinnis K, Medlin L K, Montsant A, Oudot-Le Secq M P, Napoli C, Obornik M, Parker M S, Petit J L, Porcel B M, Poulsen N, Robison M, Rychlewski L, Rynearson T A, Schmutz J, Shapiro H, Siaut M, Stanley M, Sussman M R, Taylor A R, Vardi A, von Dassow P, Vyverman W, Willis A, Wyrwicz L S, Rokhsar D S, Weissenbach J, Armbrust E V, Green B R, Van de Peer Y, Grigoriev I V. 2008. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature, 456(7219): 239-244.
DOI:10.1038/nature07410 |
Brawley S H, Blouin N A, Ficko-Blean E, Wheeler G L, Lohr M, Goodson H V, Jenkins J W, Blaby-Haas C E, Helliwell K E, Chan C X, Marriage T N, Bhattacharya D, Klein A S, Badis Y, Brodie J, Cao Y Y, Collén J, Dittami S M, Gachon C M M, Green B R, Karpowicz S J, Kim J W, Kudahl U J, Lin S J, Michel G, Mittag M, Olson B J S C, Pangilinan J L, Peng Y, Qiu H, Shu S Q, Singer J T, Smith A G, Sprecher B N, Wagner V, Wang W F, Wang Z Y, Yan J Y, Yarish C, Zäuner-Riek S, Zhuang Y Y, Zou Y, Lindquist E A, Grimwood J, Barry K W, Rokhsar D S, Schmutz J, Stiller J W, Grossman A R, Prochnik S E. 2017. Insights into the red algae and eukaryotic evolution from the genome of Porphyra umbilicalis (Bangiophyceae, Rhodophyta). Proceedings of the National Academy of Sciences of the United States of America, 114(31): E6361-E6370.
DOI:10.1073/pnas.1703088114 |
Buchfink B, Xie C, Huson D H. 2015. Fast and sensitive protein alignment using DIAMOND. Nature Methods, 12(1): 59-60.
DOI:10.1038/nmeth.3176 |
Chen N, Guo S J, Meng Q W. 2005. Relationship between plant chilling tolerance and membrane lipidscomposition and its advances in researches on molecular biology. Biotechnology Information, (2): 6-9.
(in Chinese with English abstract) |
Chin C S, Peluso P, Sedlazeck F J, Nattestad M, Concepcion G T, Clum A, Dunn C, O'Malley R, Figueroa-Balderas R, Morales-Cruz A, Cramer G R, Delledonne M, Luo C Y, Ecker J R, Cantu D, Rank D R, Schatz M C. 2016. Phased diploid genome assembly with single-molecule real-time sequencing. Nature Methods, 13(12): 1050-1054.
DOI:10.1038/nmeth.4035 |
Cock J M, Sterck L, Rouzé P, Scornet D, Allen A E, Amoutzias G, Anthouard V, Artiguenave F, Aury J M, Badger J H, Beszteri B, Billiau K, Bonnet E, Bothwell J H, Bowler C, Boyen C, Brownlee C, Carrano C J, Charrier B, Cho G Y, Coelho S M, Collén J, Corre E, Da Silva C, Delage L, Delaroque N, Dittami S M, Doulbeau S, Elias M, Farnham G, Gachon C M M, Gschloessl B, Heesch S, Jabbari K, Jubin C, Kawai H, Kimura K, Kloareg B, Küpper F C, Lang D, Le Bail A, Leblanc C, Lerouge P, Lohr M, Lopez P J, Martens C, Maumus F, Michel G, Miranda-Saavedra D, Morales J, Moreau H, Motomura T, Nagasato C, Napoli C A, Nelson D R, Nyvall-Collén P, Peters A F, Pommier C, Potin P, Poulain J, Quesneville H, Read B, Rensing S A, Ritter A, Rousvoal S, Samanta M, Samson G, Schroeder D C, Ségurens B, Strittmatter M, Tonon T, Tregear J W, Valentin K, von Dassow P, Yamagishi T, Van de Peer Y, Wincker P. 2010. The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature, 465(7298): 617-621.
DOI:10.1038/nature09016 |
Cohen P. 1976. The regulation of protein function by multisite phosphorylation. Trends in Biochemical Sciences, 1(2): 38-40.
DOI:10.1016/S0968-0004(76)80180-6 |
Collén J, Porcel B, Carré W, Ball S G, Chaparro C, Tonon T, Barbeyron T, Michel G, Noel B, Valentin K, Elias M, Artiguenave F, Arun A, Aury J M, Barbosa-Neto J F, Bothwell J H, Bouget F Y, Brillet L, Cabello-Hurtado F, Capella-Gutiérrez S, Charrier B, Cladière L, Cock J M, Coelho S M, Colleoni C, Czjzek M, Da Silva C, Delage L, Denoeud F, Deschamps P, Dittami S M, Gabaldón T, Gachon C M M, Groisillier A, Hervé C, Jabbari K, Katinka M, Kloareg B, Kowalczyk N, Labadie K, Leblanc C, Lopez P J, McLachlan D H, Meslet-Cladiere L, Moustafa A, Nehr Z, Collén P N, Panaud O, Partensky F, Poulain J, Rensing S A, Rousvoal S, Samson G, Symeonidi A, Weissenbach J, Zambounis A, Wincker P, Boyen C. 2013. Genome structure and metabolic features in the red seaweed Chondrus crispus shed light on evolution of the Archaeplastida. Proceedings of the National Academy of Sciences of the United States of America, 110(13): 5247-5252.
DOI:10.1073/pnas.1221259110 |
Curtis B A, Tanifuji G, Burki F, Gruber A, Irimia M, Maruyama S, Arias M C, Ball S G, Gile G H, Hirakawa Y, Hopkins J F, Kuo A, Rensing S A, Schmutz J, Symeonidi A, Elias M, Eveleigh R J M, Herman E K, Klute M J, Nakayama T, Oborník M, Reyes-Prieto A, Armbrust E V, Aves S J, Beiko R G, Coutinho P, Dacks J B, Durnford D G, Fast N M, Green B R, Grisdale C J, Hempel F, Henrissat B, Höppner M P, Ishida K I, Kim E, Kořený L, Kroth P G, Liu Y, Malik S B, Maier U G, McRose D, Mock T, Neilson J A D, Onodera N T, Poole A M, Pritham E J, Richards T A, Rocap G, Roy S W, Sarai C, Schaack S, Shirato S, Slamovits C H, Spencer D F, Suzuki S, Worden A Z, Zauner S, Barry K, Bell C, Bharti A K, Crow J A, Grimwood J, Kramer R, Lindquist E, Lucas S, Salamov A, McFadden G I, Lane C E, Keeling P J, Gray M W, Grigoriev I V, Archibald J M. 2012. Algal genomes reveal evolutionary mosaicism and the fate of nucleomorphs. Nature, 492(7427): 59-65.
DOI:10.1038/nature11681 |
De Clerck O, Kao S M, Bogaert K A, Blomme J, Foflonker F, Kwantes M, Vancaester E, Vanderstraeten L, Aydogdu E, Boesger J, Califano G, Charrier B, Clewes R, Del Cortona A, D'Hondt S, Fernandez-Pozo N, Gachon C M, Hanikenne M, Lattermann L, Leliaert F, Liu X J, Maggs C A, Popper Z A, Raven J A, Van Bel M, Wilhelmsson P K I, Bhattacharya D, Coates J C, Rensing S A, Van Der Straeten D, Vardi A, Sterck L, Vandepoele K, Van de Peer Y, Wichard T, Bothwell J H. 2018. Insights into the evolution of multicellularity from the sea Lettuce genome. Current Biology, 28(18): 2921-2933.e5.
|
Derelle E, Ferraz C, Rombauts S, Rouzé P, Worden A Z, Robbens S, Partensky F, Degroeve S, Echeynié S, Cooke R, Saeys Y, Wuyts J, Jabbari K, Bowler C, Panaud O, Piégu B, Ball S G, Ral J P, Bouget F Y, Piganeau G, De Baets B, Picard A, Delseny M, Demaille J, Van de Peer Y, Moreau H. 2006. Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proceedings of the National Academy of Sciences of the United States of America, 103(31): 11647-11652.
DOI:10.1073/pnas.0604795103 |
Du X M, Yin W X, Zhao Y X, Zhang H. 2001. The production and scavenging of reactive oxygen species in plants. Chinese Journal of Biotechnology, 17(2): 121-125.
(in Chinese with English abstract) |
Fan J H, Xu H, Li Y G. 2016. Transcriptome-based global analysis of gene expression in response to carbon dioxide deprivation in the green algae Chlorella pyrenoidosa. Algal Research, 16: 12-19.
DOI:10.1016/j.algal.2016.02.032 |
Fleurence J, Gutbier G, Mabeau S, Leray C. 1994. Fatty acids from 11 marine macroalgae of the French Brittany coast. Journal of Applied Phycology, 6(5-6): 527-532.
DOI:10.1007/BF02182406 |
Gao G, Zhong Z H, Zhou X H, Xu J T. 2016. Changes in morphological plasticity of Ulva prolifera under different environmental conditions: a laboratory experiment. Harmful Algae, 59: 51-58.
DOI:10.1016/j.hal.2016.09.004 |
Gao S, Chen X Y, Yi Q Q, Wang G C, Pan G H, Lin A P, Peng G. 2010. A strategy for the proliferation of Ulva prolifera, main causative species of green tides, with formation of sporangia by fragmentation. PLoS One, 5(1): e8571..
DOI:10.1371/journal.pone.0008571 |
Hanschen E R, Marriage T N, Ferris P J, Hamaji T, Toyoda A, Fujiyama A, Neme R, Noguchi H, Minakuchi Y, Suzuki M, Kawai-Toyooka H, Smith D R, Sparks H, Anderson J, Bakarić R, Luria V, Karger A, Kirschner M W, Durand P M, Michod R E, Nozaki H, Olson B J S C. 2016. The Gonium pectorale genome demonstrates co-option of cell cycle regulation during the evolution of multicellularity. Nature Communications, 7: 11370.
DOI:10.1038/ncomms11370 |
Herron M D, Desnitskiy A G, Michod R E. 2010. Evolution of developmental programs in volvox (Chlorophyta). Journal of Phycology, 46(2): 316-324.
DOI:10.1111/j.1529-8817.2009.00803.x |
Hunter T. 2007. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Molecular Cell, 28(5): 730-738.
DOI:10.1016/j.molcel.2007.11.019 |
John P C L, Mews M, Moore R. 2001. Cyclin/Cdk complexes: their involvement in cell cycle progression and mitotic division. Protoplasma, 216(3-4): 119-142.
DOI:10.1007/BF02673865 |
Joubès J, Chevalier C, Dudits D, Heberle-Bors E, Inzé D, Umeda M, Renaudin J P. 2000. CDK-related protein kinases in plants. Plant Molecular Biology, 43(5): 607-620.
|
Lagesen K, Hallin P, Rødland E A, Stærfeldt H H, Rognes T, Ussery D W. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Research, 35(9): 3100-3108.
DOI:10.1093/nar/gkm160 |
Liu G, Wang D M. 2006. The role of the plant cytoskeleton in defensing invading pathogens. Chinese Journal of Cell Biology, 28(3): 437-441.
(in Chinese with English abstract) |
Matsuzaki M, Misumi O, Shin-I T, Maruyama S, Takahara M, Miyagishima S Y, Mori T, Nishida K, Yagisawa F, Nishida K, Yoshida Y, Nishimura Y, Nakao S, Kobayashi T, Momoyama Y, Higashiyama T, Minoda A, Sano M, Nomoto H, Oishi K, Hayashi H, Ohta F, Nishizaka S, Haga S, Miura S, Morishita T, Kabeya Y, Terasawa K, Suzuki Y, Ishii Y, Asakawa S, Takano H, Ohta N, Kuroiwa H, Tanaka K, Shimizu N, Sugano S, Sato N, Nozaki H, Ogasawara N, Kohara Y, Kuroiwa T. 2004. Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature, 428(6983): 653-657.
DOI:10.1038/nature02398 |
Merchant S S, Prochnik S E, Vallon O, Harris E H, Karpowicz S J, Witman G B, Terry A, Salamov A, Fritz-Laylin L K, Maréchal-Drouard L, Marshall W F, Qu L H, Nelson D R, Sanderfoot A A, Spalding M H, Kapitonov V V, Ren Q H, Ferris P, Lindquist E, Shapiro H, Lucas S M, Grimwood J, Schmutz J, Cardol P, Cerutti H, Chanfreau G, Chen C L, Cognat V, Croft M T, Dent R, Dutcher S, Fernández E, Fukuzawa H, González-Ballester D, González-Halphen D, Hallmann A, Hanikenne M, Hippler M, Inwood W, Jabbari K, Kalanon M, Kuras R, Lefebvre P A, Lemaire S D, Lobanov A V, Lohr M, Manuell A, Meier I, Mets L, Mittag M, Mittelmeier T, Moroney J V, Moseley J, Napoli C, Nedelcu A M, Niyogi K, Novoselov S V, Paulsen I T, Pazour G, Purton S, Ral J P, Riaño-Pachón D M, Riekhof W, Rymarquis L, Schroda M, Stern D, Umen J, Willows R, Wilson N, Zimmer S L, Allmer J, Balk J, Bisova K, Chen C J, Elias M, Gendler K, Hauser C, Lamb M R, Ledford H, Long J C, Minagawa J, Page M D, Pan J M, Pootakham W, Roje S, Rose A, Stahlberg E, Terauchi A M, Yang P F, Ball S, Bowler C, Dieckmann C L, Gladyshev V N, Green P, Jorgensen R, Mayfield S, Mueller-Roeber B, Rajamani S, Sayre R T, Brokstein P, Dubchak I, Goodstein D, Hornick L, Huang Y W, Jhaveri J, Luo Y G, Martínez D, Ngau W C A, Otillar B, Poliakov A, Porter A, Szajkowski L, Werner G, Zhou K M, Grigoriev I V, Rokhsar D S, Grossman A R. 2007. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science, 318(5848): 245-250.
DOI:10.1126/science.1143609 |
Mine I, Menzel D, Okuda K. 2008. Morphogenesis in giant-celled algae. International Review of Cell and Molecular Biology, 266: 37-83.
|
Mock T, Samanta M P, Iverson V, Berthiaume C, Robison M, Holtermann K, Durkin C, BonDurant S S, Richmond K, Rodesch M, Kallas T, Huttlin E L, Cerrina F, Sussman M R, Armbrust E V. 2008. Whole-genome expression profiling of the marine diatom Thalassiosira pseudonana identifies genes involved in silicon bioprocesses. Proceedings of the National Academy of Sciences of the United States of America, 105(5): 1579-1584.
DOI:10.1073/pnas.0707946105 |
Nakamura Y, Sasaki N, Kobayashi M, Ojima N, Yasuike M, Shigenobu Y, Satomi M, Fukuma Y, Shiwaku K, Tsujimoto A, Kobayashi T, Nakayama I, Ito F, Nakajima K, Sano M, Wada T, Kuhara S, Inouye K, Gojobori T, Ikeo K. 2013. The first symbiont-free genome sequence of marine red alga, Susabi-nori (Pyropia yezoensis). PLoS One, 8(3): e57122.
DOI:10.1371/journal.pone.0057122 |
Nishitsuji K, Arimoto A, Iwai K, Sudo Y, Hisata K, Fujie M, Arakaki N, Kushiro T, Konishi T, Shinzato C, Satoh N, Shoguchi E. 2016. A draft genome of the brown alga, Cladosiphon okamuranus, S-strain: a platform for future studies of "mozuku" biology. DNA Research, 23(6): 561-570.
DOI:10.1093/dnares/dsw039 |
Ota S, Oshima K, Yamazaki T, Kim S, Yu Z, Yoshihara M, Takeda K, Takeshita T, Hirata A, Bišová K, Zachleder V, Hattori M, Kawano S. 2016. Highly efficient lipid production in the green alga Parachlorella kessleri: draft genome and transcriptome endorsed by whole-cell 3D ultrastructure. Biotechnology for Biofuels, 9(1): 13.
DOI:10.1186/s13068-016-0424-2 |
Palenik B, Grimwood J, Aerts A, Rouzé P, Salamov A, Putnam N, Dupont C, Jorgensen R, Derelle E, Rombauts S, Zhou K M, Otillar R, Merchant S S, Podell S, Gaasterland T, Napoli C, Gendler K, Manuell A, Tai V, Vallon O, Piganeau G, Jancek S, Heijde M, Jabbari K, Bowler C, Lohr M, Robbens S, Werner G, Dubchak I, Pazour G J, Ren Q H, Paulsen I, Delwiche C, Schmutz J, Rokhsar D, Van de Peer Y, Moreau H, Grigoriev I V. 2007. The tiny eukaryote Ostreococcus provides genomic insights into the paradox of plankton speciation. Proceedings of the National Academy of Sciences of the United States of America, 104(18): 7705-7710.
DOI:10.1073/pnas.0611046104 |
Polle J E W, Barry K, Cushman J, Schmutz J, Tran D, Hathwaik L T, Yim W C, Jenkins J, McKie-Krisberg Z, Prochnik S, Lindquist E, Dockter R B, Adam C, Molina H, Bunkenborg J, Jin E, Buchheim M, Magnuson J. 2017. Draft nuclear genome sequence of the Halophilic and beta-carotene-accumulating green alga Dunaliella salina strain CCAP19/18. Genome Announcements, 5(43): e01105-17.
|
Prochnik S E, Umen J, Nedelcu A M, Hallmann A, Miller S M, Nishii I, Ferris P, Kuo A, Mitros T, Fritz-Laylin L K, Hellsten U, Chapman J, Simakov O, Rensing S A, Terry A, Pangilinan J, Kapitonov V, Jurka J, Salamov A, Shapiro H, Schmutz J, Grimwood J, Lindquistz E, Lucas S, Grigoriev I V, Schmitt R, Kirk D, Rokhsar D S. 2010. Genomic analysis of organismal complexity in the multicellular green alga Volvox carteri. Science, 329(5988): 223-226.
DOI:10.1126/science.1188800 |
Saxena I, Srikanth S, Chen Z. 2016. Cross talk between H2O2 and interacting signal molecules under plant stress response. Frontiers in Plant Science, 7: 570.
|
Schattner P, Brooks A N, Lowe T M. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Research, 33(S2): W686-W689.
|
Sies H. 1993. Strategies of antioxidant defense. European Journal of Biochemistry, 215(2): 213-219.
DOI:10.1111/j.1432-1033.1993.tb18025.x |
Simão F A, Waterhouse R M, Ioannidis P, Kriventseva E V, Zdobnov E M. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31(19): 3210-3212.
DOI:10.1093/bioinformatics/btv351 |
Stanke M, Steinkamp R, Waack S, Morgenstern B. 2004. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Research, 32(S2): W309-W312.
|
Tan I H, Blomster J, Hansen G, Leskinen E, Maggs C A, Mann D G, Sluiman H J, Stanhope M J. 1999. Molecular phylogenetic evidence for a reversible morphogenetic switch controlling the gross morphology of two common genera of green seaweeds, Ulva and Enteromorpha. Molecular Biology & Evolution, 16(8): 1011-1018.
|
Temporini C, Calleri E, Massolini G, Caccialanza G. 2008. Integrated analytical strategies for the study of phosphorylation and glycosylation in proteins. Mass Spectrometry Reviews, 27(3): 207-23.
DOI:10.1002/mas.20164 |
Wickstead B, Ersfeld K, Gull K. 2003. The mitotic stability of the minichromosomes of Trypanosoma brucei. Molecular & Biochemical Parasitology, 132(2): 97-100.
|
Worden A Z, Lee J H, Mock T, Rouzé P, Simmons M P, Aerts A L, Allen A E, Cuvelier M L, Derelle E, Everett M V, Foulon E, Grimwood J, Gundlach H, Henrissat B, Napoli C, McDonald S M, Parker M S, Rombauts S, Salamov A, Von Dassow P, Badger J H, Coutinho P M, Demir E, Dubchak I, Gentemann C, Eikrem W, Gready J E, John U, Lanier W, Lindquist E A, Lucas S, Mayer K F X, Moreau H, Not F, Otillar R, Panaud O, Pangilinan J, Paulsen I, Piegu B, Poliakov A, Robbens S, Schmutz J, Toulza E, Wyss T, Zelensky S, Zhou K M, Armbrust E V, Bhattacharya D, Goodenough U W, Van de Peer Y, Grigoriev I V. 2009. Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes Micromonas. Science, 324(5924): 268-272.
DOI:10.1126/science.1167222 |
Wu H L, Gao G, Zhong Z H, Li X S, Xu J T. 2018. Physiological acclimation of the green tidal alga Ulva prolifera to a fast-changing environment. Marine Environmental Research, 137: 1-7.
DOI:10.1016/j.marenvres.2018.02.018 |
Xu D L, Huang X C, Yang W G, Wu D, Cao W Q. 2003. Analysis of nutrition composition of Enteromorpha prolifera. Journal of Zhejiang Ocean University (Natural Science), 22(4): 318-320.
(in Chinese with English abstract) |
Yang X J. 2005. Multisite protein modification and intramolecular signaling. Oncogene, 24(10): 1653-1662.
DOI:10.1038/sj.onc.1208173 |
Ye N H, Zhang X W, Miao M, Fan X, Zheng Y, Xu D, Wang J F, Zhou L, Wang D S, Gao Y, Wang Y T, Shi W Y, Ji P F, Li D M, Guan Z, Shao C W, Zhuang Z M, Gao Z Q, Qi J, Zhao F Q. 2015. Saccharina genomes provide novel insight into kelp biology. Nature Communications, 6(1): 6986-6996.
DOI:10.1038/ncomms7986 |
Zelko I N, Mariani T J, Folz R J. 2002. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radical Biology and Medicine, 33(3): 337-349.
DOI:10.1016/S0891-5849(02)00905-X |
Zhang K S, Tian H L. 2007. Research and function of catalase in organism. Food Science & Technology, 32(1): 8-11.
(in Chinese with English abstract) |