 2021, Vol. 39
2021, Vol. 39Institute of Oceanology, Chinese Academy of Sciences
Article Information
- WANG Junhao, MAO Yunxiang, DU Guoying, LI Xiaojiao, TANG Xianghai
- On microbial community of Pyropia haitanensis by metagenomic analysis
- Journal of Oceanology and Limnology, 39(3): 1091-1102
- http://dx.doi.org/10.1007/s00343-020-0189-0
Article History
- Received May. 10, 2020
- accepted in principle May. 23, 2020
- accepted for publication Jun. 4, 2020




2 Key Laboratory of Utilization and Conservation of Tropical Marine Bioresource(Ministry of Education), College of Fisheries and Life Science, Hainan Tropical Ocean University, Sanya 572022, China
Pyropia is an important commercial product that is widely cultivated in the south coastal areas of China (Xu et al., 2015). Previous studies have shown that the growth status of Pyropia is closely related to its microbial diversity. Several researches have focused on the microbial diversity of Pyropia by using marker genes such as 16S rRNA genes, 18S rRNA genes, and internal transcribed spacer (ITS) genes (Yang et al., 2008; Shen et al., 2013). A recent study investigated the shift of Pyropia-associated bacterial communities by using 16S rRNA gene sequencing and showed that there were more than 300 operational taxonomic units (OTUs) in their samples, distributed in approximately 15 microbial phyla (Yan et al., 2019). The microbial composition of Pyropia, whether healthy or diseased, is complex. Other similar studies investigated the microbial community composition of Pyropia using amplicon sequencing. However, owing to host contamination, PCR biases may appear in the amplification of 16S rRNA gene.
In this study, to obtain a more accurate microbial information, we performed taxonomic profiling based on shotgun metagenome sequencing of whole community DNA, which provides a comparatively unbiased insight into the microbial composition of P. haitanensis. In addition, as metagenomics examined the entire genetic material rather than the identification sequences (e.g., 16S rRNA) (Ngara and Zhang, 2018; Shi et al., 2019), metagenome sequencing technology can solve the complex functional problems of microorganisms (Chen et al., 2019; Qiu et al., 2019). The overall functional analysis of the microbial community of P. haitanensis revealed functional genes for the synthesis of vitamin B12 and indole-3-acetic acid (IAA). Vitamin B12 plays an essential role in many biosynthetic pathways. However, the pathway involved in the synthesis of vitamin B12 has been found only in prokaryotes (Bertrand et al., 2011), such as Pseudomonas denitrificans (Warren et al., 2002), Salmonella typhimurium (Roth et al., 1993), and Bacillus megaterium (Raux et al., 1998; Cruz-López and Maske, 2016). The production of the phytohormone IAA is considered a major plant-growth-promoting (PGP) feature of plant-beneficial bacteria (Bulgarelli et al., 2013; Nelkner et al., 2019).
In a previous study, 15 uncultured microbial genomes were obtained from bovine rumen by binning method (Hess et al., 2011). Similarly, specific information on the microbial species and genomes in the microbial community of P. haitanensis should be determined. Therefore, in this study, metagenomic analysis was performed to identify the microbial community composition and function of microorganisms of P. haitanensis. We identified six high-quality metagenome-assembled genomes (MAGs) associated with the synthesis of vitamin B12 and IAA by binning method. In our study, we used metagenomics to analyze the microbial community structure and function of the microorganism community of P. haitanensis, which enriched the information of microflora structure, fills in the blank of microflora function, and provided a new research idea for the study of laver symbiotic microbe.
2 MATERIAL AND METHOD 2.1 Experimental material of P. haitanensisThe gametophytes of P. haitanensis (PH 40) were obtained from a laboratory culture from the Laboratory of Phycological Genetics and Sometic Cell Engineering, Ocean University of China. The gametophytes were continuously cultivated with Provasoli's enrichment solution medium at 20±3 ℃ and light intensity of 20 μmol photons/(m2s) following a 12-h light and 12-h dark cycle. DNA extraction was performed when the length of a healthy gametophyte was approximately 8-10 cm.
2.2 DNA extraction, library construction, and sequencingGametophytes with a length of around 8-10 cm were selected to extract genomic DNA according to the instructions of Plant Genomic DNA Kit, TIANGEN Biotech (Beijing) Co., Ltd. The concentration and quality (A260/A280) of extracted DNA were determined using a NanoDrop ND-2000 spectrophotometer (NanoDrop, Wilmington, DE, USA), and evaluated with a 1% agarose gel. To minimize DNA extraction bias, three replicate DNA isolations were pooled (Feng et al., 2018). Pair-end library building and sequencing were completed at Personal Biotechnology Co., Ltd. (Shanghai, China) according to the standard protocol (http://www.illumina.com/).
2.3 Preprocessing of sequencing dataTrim Galore (V0.5.0) was used to remove adapter and low quality reads on returned data from the company (Gdula et al., 2019). The reads that were compared with the P. haitanensis genome (including plastids and mitochondrial genomes) were removed using Bowtie2 (V.2.3.2) (Langmead and Salzberg, 2012; Cao et al., 2020). PCR repeats were removed using FastUnique (V.1.1) (Xu et al., 2012). The clean reads were matched back to the host genome to estimate contamination from host genome. To obtain the contigs dataset, metaSPAdes (V.3.9.0) was used to perform the assembly of clean reads after quality control (Bankevich et al., 2012). Then, QUAST (V.4.5, http://quast.bioinf.spbau.ru/) was used to obtain the statistics for all assemblies.
2.4 Taxonomy assignment, gene prediction, and functional annotationThe taxonomic assignment of metagenomes was performed with KAIJU (http://kaiju.binf.ku.dk/server) using the default parameters. The taxonomic annotation of microorganisms was obtained by inhouse shell scripts, and species abundance was calculated by the reads number. The open reading frames (ORFs) were predicted through Prodigal (V.2.6.3) (Hyatt et al., 2012). The unique gene dataset was obtained using CD-HIT (V.4.8.1) to remove redundant genes (Fu et al., 2012). Then, the ORFs were aligned using the eggNOG database (evolutionary genealogy of genes: Non-supervised Orthologous Groups, Version 4.0) via eggNOG mapper (V.0.3) with the default parameters (Huerta-Cepas et al., 2016), and the corresponding cluster of orthologous groups of protein (COG) was obtained. The KAAS (KEGG Automatic Annotation Server) (Moriya et al., 2007) program was used to predict KEGG pathway annotations by performing GHOSTX search and SBH method against the Kyoto Encyclopedia of Genes and Genomes database (KEGG GENES). Carbohydrate-active enzymes (CAZymes) for sequences of protein with more than 100 bp were annotated by using the dbCAN2 (V7.0) website (Zhang et al., 2018). The software MetaWRAP (V.1.0.5) was used to obtain MAGs by binning method (Uritskiy et al., 2018). The completeness and contamination of MAGs were estimated by CheckM.
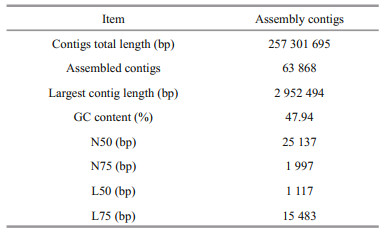
3 RESULT 3.1 Sequencing results and data preprocessingThe original data were finally obtained 28 Gb clean data after quality control. In total, 114 086 645 reads were obtained, and the proportion of reads with base quality score greater than 99% (Quality score > 20) was 100%. The total alignment rate between clean reads and host genome was less than 0.01%. The SPAdes software was used to assemble data (meta parameters) after quality control, and the fragments with length less than 1 000 bp in the assembly results were removed. After gene prediction and redundancy removal, 63 868 contig sequences and 241 293 genes were obtained. Then, clean reads were mapped back to the contigs, and the reads utilization rate was 90.69%. The results of assembly evaluation by QUAST are presented in Table 1.
The clean reads were used to query against the taxonomic database (RefSeq non-redundant proteins database, NR) and only the reads with a bit score > 75 were extracted for the following analysis. A total of 51 453 077 (45.1%) reads were assigned to the taxonomic database (Fig. 1). Sequences annotated to the bacterial community accounted for 98.98% and constituted the main part of the microbiota. In addition, eukaryotes, archaea, and viruses accounted for 0.8%, 0.2%, and 0.02% of the microbiota, respectively.

|
| Fig.1 Relative abundance of phylum, genus, and species in microorganisms Sankey diagram showing the relative abundance of species at three taxonomic levels, in which the phylum level is higher than 0.1%, the genus level higher than 1%, and the species level higher than 1% are shown. The curve shows the mapping of phylum, genus, and species. The thickness indicates the relative abundance. |
At the phylum level, 171 taxa were obtained; five taxa had an abundance of more than 1%, namely Proteobacteria (54.64%), Bacteroidetes (37.92%), Actinomycetes (1.30%), Fusarium (1.35%), and Firmicutes (1.45%). The relative abundance of 14 taxa was higher than 0.1%, which included Basidiomycota (0.1%) and Ascomycota (0.1%).
At the genus level, 3 159 genera were obtained; 12 genera had a horizontal abundance of more than 1%, 120 genera had an abundance of more than 0.1%, and 580 genera had an abundance of more than 0.01%. The relative abundance of 12 genera was greater than 1%, of which Erythrobacter (3.98%), Sphingorhabdus (2.46%), Sulfitobacter (2.37%), Altererythrobacter (1.84%), Marinobacter (1.12%), Hoeflea (1.08%), and Labrenzia (1.56%) belonged to Proteobacteria; and Lewinella (1.94%), Hyunsoonleella (1.56%), Aquimarina (1.16%), Flavobacterium (1.01%), and Jejuia (1.01%) belonged to Bacteroidetes. At the species level, 16 691 species were obtained. The relative abundance of four species was greater than 1%, of which Hyunsoonleella jejuensis (1.56%) and Jejuia pallidilutea (1.01%) belonged to Bacteroidetes; and Sphingorhabdus marina (1.48%) and Sphingomonadales bacterium EhC05 (1.48%) belonged to Proteobacteria. The relative abundance of 91 species was more than 0.1% and that of 1 134 species was more than 0.01%.
3.3 COG annotationThe eggNOG mapper was used to map protein sequences to the eggNOG database by running the diamond mode. Notably, 30.95% of the predicted genes identified in the dataset were assigned to putative functions (Fig. 2). The classification of potential genes was subsequently conducted by COG analysis.

|
| Fig.2 Distribution of predicted genes in the COG classification "A-W", "Y", and "Z" stand for COG categories. Different colors represent different classes. "Unclass" means the genes that had not been classified; those with fewer than 100 genes were defined as "others". |
General function prediction only [R] was the dominant function among the 25 categories, followed by Amino acid transport and metabolism [E], Transcription [K], Signal transduction mechanisms [T], Carbohydrate transport and metabolism [G], and Cell wall/membrane/envelope biogenesis [M] (> 9 000). The lowest number of genes (< 65) were assigned to RNA processing and modification [A], Chromatin structure and dynamics [B], Nuclear structure [Y], and Cytoskeleton [Z]. In addition, when the potential functional genes were matched to the NCBI Taxonomy Database, we found Proteobacteria (Alphaproteobacteria in particular) was the major phylum in every COG category, which consistent with species abundance distribution.
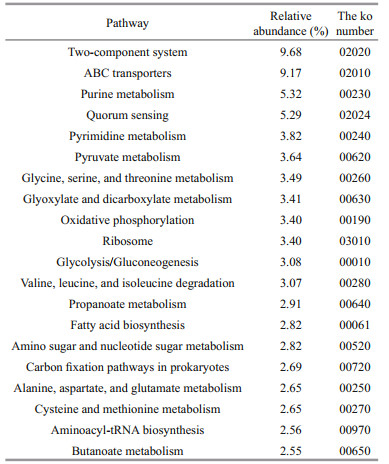
3.4 KEGG function annotationIn addition to gene function insights, metagenomic analysis can also provide an opportunity to understand high-level functions and utilities of the microbial community of P. haitanensis. In total, we identified 90 247 KOs (KEGG orthologys), 417 pathways, and 130 modules. Sixty-nine pathways with a relative abundance greater than 1.00% were obtained, and they were defined as dominant pathways. The ko02010 was the most abundant KEGG pathway, which was assigned to ABC transporters (Table 2), followed by two-component system and quorum sensing. There were many pathways of amino acid biosynthesis and metabolism, such as tyrosine, lysine, histidine, valine, leucine, isoleucine, cysteine, methionine, alanine, aspartate, and glutamate. The enzymes of the KEGG Tryptophan Metabolism (map 00380) pathway potentially involved in indole-acetic acid (IAA, auxin) biosynthesis were identified. In particular, the genes for aldehyde dehydrogenase (NAD+), amidase, and monoamine oxidase were present, implying the presence of auxin synthesized by the tryptamine pathway. The miaA enzyme (K00791) gene capable of synthesizing cis-zeatin was identified in the carotenoid biosynthesis pathway (ko00906), and P-carotene produced in this pathway which was the precursor for biosynthesis of vitamin A. Regarding vitamin metabolism, various complete functional units in the bacterial community encoded vitamins, including thiamine biosynthesis (M00127), pyridoxal biosynthesis (M00124), pantothenic acid biosynthesis (M00119), biotin biosynthesis (M00123), cobalamin biosynthesis (M00122), and methylnaphthoquinone biosynthesis (M00116).
The Carbohydrate-Active enZYmes Database (CAZy database) can be divided into six categories: glycosyl transferases (GTs), glycoside hydrolases (GHs), auxiliary activities (AAs), carbohydrate esterases (CEs), proteins with carbohydrate-binding modules (CBMs) and polysaccharide lyases (PLs).
After annotation (e-value < 1e-5, coverage > 0.35), 4 273 genes were annotated by dbCAN2 against CAZy database. GTs (37.22%) and GHs (34.28%) were the dominant CAZyme categories, followed by CEs (15.23%), AAs (5.99%), CBMs (5%), and PLs (2.28%) (Fig. 3). In total, 1 591 genes were distributed in different GT families, with Proteobacteria and Bacteroidetes accounting for 64.3%, and 27% of the GT family, respectively. Epibacterium mobile F1926 (4%), Hyphomonas sp. Mor2 (3.6%), and Methylotenera versatilis 301 (3.35%) were the top three bacterial strains in the GT families. GT2 (33.60%), GT4 (24.51%), and GT51 (10.67%) were the three most abundant GTs containing multiple enzymes related to cell wall synthesis. GH was the second dominant category; 1 465 genes were distributed in different GH families, of which Proteobacteria and Bacteroidetes accounted for 50.9% and 37.7% of the GH family, respectively. Seonamhaeicola sp. S2-3 (4.7%), Aquimarina sp. AD10 (4.6%), and Algibacter alginicilyticus (3%) were top three bacterial strains in the GH family. GH13 (9.33%) and GH23 (9.12%) were the dominant GHs. GH13 is a big family and includes more subfamilies such as hydrolases, transglycosidases, and isomerases (Svensson, 1994; Janeček, 1997; MacGregor et al., 2001). Branching enzyme (Subf 9) was the most abundant enzyme in all GH13 subfamilies, followed by a-glucosidase (Subf 23). The former can convert amylose into amylopectin, while the latter can hydrolyze oligosaccharides rapidly (Bruni et al., 1970). GH23 included lysozyme and chitinase, which could be a possible way by which microbes compete. We also identified the family of enzymes that can degrade agarose, including GH16, GH50, GH86, and GH118. The relative abundance of CEs, AAs, CBMs, and PLs was lower than the above two categories. CE10 was rank one in Carbohydrate Esterase family; however, majority of the members of this family are esterases acting on non-carbohydrate substrates, included arylesterase, carboxyl esterase, acetylcholinesterase, cholinesterase, sterol esterase, and brefeldin A esterase. AA3 family was the most abundant in the auxiliary activity family, and it belonged to the glucose-methanol-choline oxidoreductase family. CBM9 was the most abundant family in the carbohydrate-binding module family, and it has been known as cellulose-binding domain family IX. In the polysaccharide lyase superfamily, PL6 and PL7 were the families with the highest relative abundance. Thirteen PL6 proteins were not classified to any subfamily, and nine proteins were classified to alginate lyase (Subf 1). Similar to the PL6 family, 58% of PL7 proteins were classified as alginate lyase (Subf 5). Furthermore, Alphaproteobacteria was a major class in GT family, AA family, and CE family, while Flavobacteriia was higher abundance in the other CAZymes categories.

|
| Fig.3 Distribution of predicted genes in CAZyme classification The six CAZyme categories are shown on the horizontal axis; the number of genes on the vertical axis. Different colors represent class levels. "Unclass" means the genes that had not been classified. |
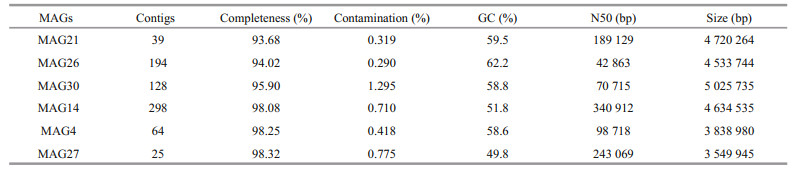
Several essential genes coding for enzymes involved in vitamin B12 biosynthesis were used to predict the MAGs with potential to synthesize cobalamin, including cbiA/cobB encoding cobyrinic acid a, c-diamide synthase, cbiC/cobH encoding precorrin-8x methylmutase, and cobT encoding nicotinate mononucleotide: 5, 6-dimethylbenzimi-dazole phosphoribosyltransferase. Each of these genes represents a potential biomarker for vitamin biosynthesis (Bertrand et al., 2011). When these genes were present in fully sequenced bacterial and archaeal genomes, the complete B12 biosynthesis pathway was also present; the genes were homologous in both oxygen-requiring (cobB, cobH, and cobT) and nonoxygen-requiring pathways for vitamin synthesis (cbiA, cbiC, and cobT) (Bertrand et al., 2011). Finally, six MAGs were identified as containing the above essential genes (Supplementary Table S1): MAG4, MAG14, MAG21, MAG26, MAG27, MAG30 and the results of MAGs evaluation by CheckM are presented in Table 3.
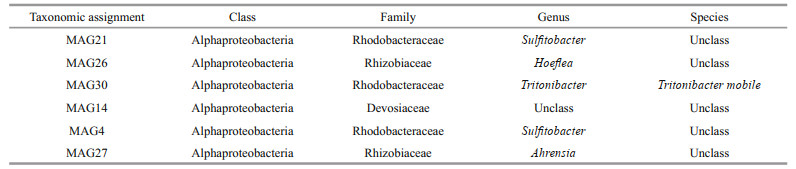
Then, we obtained taxonomic information for the six MAGs based on the Genome Taxonomy Database (GTDB, 04-RS89) by GTDB-Tk (V.0.3.3) (Table 4). The result shows that all the six MAGs belonged to Alphaproteobacteria, which included Rhodobacteraceae, Rhizobiaceae, and Devosiaceae. Only MAG30 was assigned to species level with close genetic distance with Epibacterium mobile (GCF_001681715.1, ANI=96.77%).
The KEGG annotation results were searched for IAA-related enzymes in all MAGs, and a complete pathway (tryptamine pathway) for IAA synthesis was found in the MAG21 draft genome. The IAA synthesis pathway was involved in tryptophan metabolism (ko00380) pathway; the related enzyme gene information is shown in Table 5.

In this study, we aimed to determine the function of the microbial community of P. haitanensis through metagenomic analysis for the first time. We obtained metagenome sequence data and assembled the gene sets that represent a valuable reference repository, particularly for Pyropia. After obtaining genetic information of all microorganisms in the sample by metagenome sequencing, we investigated the structure and functional potential of the microbial community of P. haitanensis (Sudarikov et al., 2017).
A stable microbial community is a key factor in maintaining the growth and development of P. haitanensis. By characterizing the abundance of reads at the phylum, genera, and species levels, metagenome sequencing revealed the microbial abundance and diversity of the gametophytes of P. haitanensis. Twelve phyla with abundance greater than 0.1% were obtained. Among them, Proteobacteria (54.64%) and Bacteroidetes (37.92%) were the dominant phyla, which comprised more than 75% of the total population. We reviewed 161 macroalgal-bacterial studies over the past few decades, and a bacterial core community comprising Proteobacteria (especially Alphaproteobacteria and Gammaproteobacteria), CFB group, Firmicutes, and Actinobacteria species was found to be functionally closely related to the host (Cruz-López and Maske, 2016). The result showed that the algal microorganisms were similar to some extent. Therefore, we speculate that Proteobacteria and Bacteroidetes may have important influence on the growth and development of P. haitanensis. Erythrobacter was the most abundant genus. They can degrade alkanes, oxidize tellurite, and form tellurite crystals to decrease the concentration of tellurite acid compounds in the environment and reduce the biological toxicity of tellurite acid compounds (Yurkov et al., 1996; Alonso-Gutiérrez et al., 2009). In addition, Erythrobacter had a strong production capacity of astaxanthin and carotenoids and played an important role in the global ocean carbon cycle and energy metabolism (Noguchi et al., 1992). We detected bacteria that caused Pyropia disease, such as Cobetia marina, Fusarium sp., Pseudoalteromonas citrea, and Pseudoalteromonas tetraodonis, but their abundance was extremely low (< 0.001%). Yang et al. (2008) proposed that the microorganisms of Pyropia, such as Marinobacter, Planococcus, and Macrococcus, may be regional. However, in our analysis, these three types of bacteria were detected, indicating that the differences in microorganisms between regions may be differences in terms of abundance rather than species. The present study also showed that although the abundance of Pseudomonas, which is associated with Pyropia health, was higher than 0.1%, it was not the dominant species. In addition, virus sequences were found in the data (0.02%), among which Caudovirales was the most abundant, accounting for approximately 62.3% of the total virus sequences. Although there were pathogenic bacteria and viruses among microorganisms of P. haitanensis, their abundance was very low, and they do not necessarily cause algal disease (Feng et al., 2018).
The gametophytes of microorganisms were quite abundant, and many of them had numerous physiological functions. Nelkner et al. (2019) found that Amino acid transport and metabolism (E) was ranked two in terms of abundance (category R was on rank one) in soil microorganisms, and amino acid metabolism may be of key importance for the soil microbiome analyzed. In the present study, we found that most of the functional genes were involved in microbial metabolism, and Amino acid transport and metabolism (E) was ranked two (category R was on rank one) among the 25 functional categories. These findings indicated that amino acids might play an important role between gametophytes and host. On the other hand, the results show that although the rhizospheric microorganism and P. haitanensis microorganism were in different environmental media, the corresponding functions of the microbial community were universal to a certain extent.
The most abundant KEGG pathway was ko02010, which was assigned to ABC transporter. The current research on microbial ABC transporters showed that they were involved in many biological functions, such as transport of ions, amino acids, nucleotides, polysaccharides, and peptides; bacterial drug resistance; pheromone secretion; and detoxification of heavy metals (Dean and Annilo, 2005; Davidson et al., 2008; Theodoulou and Kerr, 2015). It was also reported that ABC transporter played an important role in plants (Do et al., 2018). The other abundant KEGG pathways were two-component system and quorum sensing, which play important roles in adaptive mechanisms of microorganisms to the environment, such as colonization, nutrient acquisition, and collective defense (Kleerebezem et al., 1997; Hmelo, 2017).
The abundance of GT was higher than that of GH. The abundance of CEs, AAs, CBMs, and PLs was lower than that of GHs. Among all GHs, amylase GH13 (9.33%) and lysozyme GH23 (9.12%) were the most abundant CAZymes. We also found that the presence of agarase and alginate lyase from the GH family and PL family, which indicated that the host might provide diverse carbon sources for the microflora. Lysozyme usually plays a role in maintaining the stability of the microflora (Li et al., 2019).
Most carbohydrate enzymes belonged to Proteobacteria and Bacteroidetes (especially Alphaproteobacteria and Flavobacteriia), implying that Proteobacteria not only play an important role in species abundance, but also play a pivotal role in function.
Metagenome assembly and binning were performed to reconstruct genomes of unknown and abundant microbial community members. By using binning method, six MAGs were obtained, including MAG4, MAG14, MAG21, MAG26, MAG27, and MAG30, which are potentially involved in cobalamin biosynthesis and IAA synthesis. Vitamin B12, a structurally complex and functionally important vitamin, is one of the essential vitamins required for the growth of P. haitanensis. It has been reported that Lingulodinium polyedrum is a vitamin B1 and B12 auxotroph and may acquire both vitamins from the associated bacterial community, especially Proteobacteria having high abundance (Croft et al., 2005). Similarly, P. haitanensis was auxotrophic for vitamin B12 (Croft et al., 2005; Bertrand et al., 2011). The fact that vitamin B12 can only be formed by bacteria and archaea implies that vitamin B12-producing microorganisms play an important role in P. haitanensis (Watanabe, 2007; Bertrand et al., 2011; Helliwell, 2017; Wichard and Beemelmanns, 2018). On the other hand, bacteria that produce IAA were generally considered PGP microbiome members (Bulgarelli et al., 2013; Nelkner et al., 2019). By putting the whole draft genomes to GTDB, we speculated MAGs as new strains in Rhodobacteraceae, Rhizobiaceae, and Devosiaceae. All MAGs with completeness > 90% and contamination < 1.5% can provide a strategy for the isolation, culture, and functional identification of microorganisms of P. haitanensis.
Under the influence of experimental materials and sequencing data, we conducted a systematic study on the microflora of gametophytes of P. haitanensis, and a deeper study on the complex relationship between host and microflora will be reflected in subsequent studies.
5 CONCLUSIONThe microbial community of includes not only prokaryotes but also fungi and viruses or bacteriophages. In this study, we comprehensively analyzed the microbial species diversity of P. haitanensis and systematically analyzed their functions. We found six MAGs associated with the synthesis of vitamin B12 and IAA, and they can added to the microorganism genome database of P. haitanensis. The obtained genome information for the new candidate P. haitanensis beneficial microbial species may guide the development of rational isolation strategies. We used metagenomic to analysis the microorganism community of P. haitanensis, which enriched the information of microflora structure and function, and provided a new research idea for investigating laver symbiotic microbe.
6 DATA AVAILABILITY STATEMENTThe datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Electronic supplementary materialSupplementary material (Supplementary Table S1) is available in the online version of this article at https://doi.org/10.1007/s00343-020-0189-0.
Alonso-Gutiérrez J, Figueras A, Albaigés J, Jiménez N, Viñas M, Solanas A M, Novoa B. 2009. Bacterial communities from shoreline environments (Costa da Morte, Northwestern Spain) affected by the Prestige oil spill. Applied and Environmental Microbiology, 75(11): 3 407-3 418.
DOI:10.1128/AEM.01776-08 |
Bankevich A, Nurk S, Antipov D, Gurevich A A, Dvorkin M, Kulikov A S, Lesin V M, Nikolenko S I, Pham S, Prjibelski A D, Pyshkin A V, Sirotkin A V, Vyahhi N, Tesler G, Alekseyev M A, Pevzner P A. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology, 19(5): 455-477.
DOI:10.1089/cmb.2012.0021 |
Bertrand E M, Saito MA, Jeon Y J, Neilan B A. 2011. Vitamin B12 biosynthesis gene diversity in the Ross Sea: the identification of a new group of putative polar B12 biosynthesizers. Environmental Microbiology, 13(5): 1 285-1 298.
DOI:10.1111/j.1462-2920.2011.02428.x |
Bruni C B, Sica V, Auricchio F, Covelli I. 1970. Further kinetic and structural characterization of the lysosomal α-D-glucoside glucohydrolase from cattle liver. Biochimica et Biophysica Acta (BBA) - Enzymology, 212(3): 470-477.
DOI:10.1016/0005-2744(70)90253-6 |
Bulgarelli D, Schlaeppi K, Spaepen S, van Themaat E V L, Schulze-Lefert P. 2013. Structure and functions of the bacterial microbiota of plants. Annual Review of Plant Biology, 64: 807-838.
DOI:10.1146/annurev-arplant-050312-120106 |
Cao M, Xu K P, Yu X Z, Bi G Q, Liu Y, Kong F N, Sun P P, Tang X H, Du G Y, Ge Y, Wang D M, Mao Y X. 2020. A chromosome-level genome assembly of Pyropia haitanensis (Bangiales, Rhodophyta). Molecular Ecology Resources, 20(1): 216-227.
DOI:10.1111/1755-0998.13102 |
Chen L, Fang K, Zhou J, Yang Z P, Dong X F, Dai G H, Zhang H B. 2019. Enrichment of soil rare bacteria in root by an invasive plant Ageratina adenophora. Science of the Total Environment, 683: 202-209.
DOI:10.1016/j.scitotenv.2019.05.220 |
Croft M T, Lawrence A D, Raux-Deery E, Warren M J, Smith A G. 2005. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature, 438(7064): 90-93.
DOI:10.1038/nature04056 |
Cruz-López R, Maske H. 2016. The Vitamin B1 and B12 required by the marine Dinoflagellate lingulodinium polyedrum can be provided by its associated bacterial community in culture. Frontiers in Microbiology, 7: 560.
DOI:10.3389/fmicb.2016.00560 |
Davidson A L, Dassa E, Orelle C, Chen J. 2008. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiology and Molecular Biology Reviews, 72(2): 317-364.
DOI:10.1128/MMBR.00031-07 |
Dean M, Annilo T. 2005. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annual Review of Genomics and Human Genetics, 6: 123-142.
DOI:10.1146/annurev.genom.6.080604.162122 |
Do T H T, Martinoia E, Lee Y. 2018. Functions of ABC transporters in plant growth and development. Current Opinion in Plant Biology, 41: 32-38.
DOI:10.1016/j.pbi.2017.08.003 |
Feng G, Xie T, Wang X, Bai J Y, Tang L, Zhao H, Wei W, Wang M L, Zhao Y. 2018. Metagenomic analysis of microbial community and function involved in cd-contaminated soil. BMC Microbiology, 18(1): 11.
DOI:10.1186/s12866-018-1152-5 |
Fu L M, Niu B F, Zhu Z W, Wu S T, Li W Z. 2012. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28(23): 3 150-3 152.
DOI:10.1093/bioinformatics/bts565 |
Gdula M R, Nesterova T B, Pintacuda G, Godwin J, Zhan Y, Ozadam H, McClellan M, Moralli D, Krueger F, Green C M, Reik W, Kriaucionis S, Heard E, Dekker J, Brockdorff N. 2019. The non-canonical SMC protein SmcHD1 antagonises TAD formation and compartmentalisation on the inactive X chromosome. Nature Communications, 10(1): 30.
DOI:10.1038/s41467-018-07907-2 |
Helliwell K E. 2017. The roles of B vitamins in phytoplankton nutrition: new perspectives and prospects. New Phytologist, 216(1): 62-68.
DOI:10.1111/nph.14669 |
Hess M, Sczyrba A, Egan R, Kim T W, Chokhawala H, Schroth G, Luo S J, Clark D S, Chen F, Zhang T, Mackie R I, Pennacchio L A, Tringe S G, Visel A, Woyke T, Wang Z, Rubin E M. 2011. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science, 331(6016): 463-467.
DOI:10.1126/science.1200387 |
Hmelo L R. 2017. Quorum sensing in marine microbial environments. Annual Review of Marine Science, 9: 257-281.
DOI:10.1146/annurev-marine-010816-060656 |
Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter M C, Rattei T, Mende D R, Sunagawa S, Kuhn M, Jensen L J, von Mering C, Bork P. 2016. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Research, 44(D1): D286-D293.
DOI:10.1093/nar/gkv1248 |
Hyatt D, LoCascio P F, Hauser L J, Uberbacher E C. 2012. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics, 28(17): 2 223-2 230.
DOI:10.1093/bioinformatics/bts429 |
Janeček S. 1997. α-amylase family: molecular biology and evolution. Progress in Biophysics and Molecular Biology, 67(1): 67-97.
DOI:10.1016/S0079-6107(97)00015-1 |
Kleerebezem M, Quadri L E, Kuipers O P, de Vos W M. 1997. Quorum sensing by peptide pheromones and two-component signal-transduction systems in Gram-positive bacteria. Molecular Microbiology, 24(5): 895-904.
DOI:10.1046/j.1365-2958.1997.4251782.x |
Langmead B, Salzberg S L. 2012. Fast gapped-read alignment with Bowtie 2. Nature Methods, 9(4): 357-359.
DOI:10.1038/nmeth.1923 |
Li T T, Qi M T, Gatesoupe F J, Tian D C, Jin W H, Li J, Lin Q, Wu S J, Li H. 2019. Adaptation to fasting in crucian carp (Carassius auratus): gut microbiota and its correlative relationship with immune function. Microbial Ecology, 78(1): 6-19.
DOI:10.1007/s00248-018-1275-0 |
Ma H X, Zhang L L, Sun X M, Sun H Q, He M X, Chen G J, Wang L S. 2015. Understanding microbial communities and their functions by meta-omics approaches. Microbiology China, 42(5): 902-912.
(in Chinese with English abstract) DOI:10.13344/j.microbiol.china.140965 |
MacGregor E A, Janeček Š, Svensson B. 2001. Relationship of sequence and structure to specificity in the α-amylase family of enzymes. Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology, 1546(1): 1-20.
DOI:10.1016/S0167-4838(00)00302-2 |
Moriya Y, Itoh M, Okuda S, Yoshizawa A C, Kanehisa M. 2007. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Research, 35(suppl_2): W182-W185.
DOI:10.1093/nar/gkm321 |
Nelkner J, Henke C, Lin T W, Pätzold W, Hassa J, Jaenicke S, Grosch R, Pühler A, Sczyrba A, Schlüter A. 2019. Effect of long-term farming practices on agricultural soil microbiome members represented by metagenomically assembled genomes (MAGs) and their predicted plant-beneficial genes. Genes, 10(6): 424.
DOI:10.3390/genes10060424 |
Ngara T R, Zhang H J. 2018. Recent advances in function-based metagenomic screening. Genomics, Proteomics & Bioinformatics, 16(6): 405-415.
DOI:10.1016/j.gpb.2018.01.002 |
Noguchi T, Hayashi H, Shimada K, Takaichi S, Tasumi M. 1992. In vivo states and functions of carotenoids in an aerobic photosynthetic bacterium, Erythrobacter longus. Photosynthesis Research, 31(1): 21-30.
DOI:10.1007/BF00049533 |
Qiu M, Huang K Q, Liu Y Z, Yang A Y, Tang H L, Liu X X, Wang C L, Chen H L, Xiong Y, Zhang J, Yang J. 2019. Modulation of intestinal microbiota by glycyrrhizic acid prevents high-fat diet-enhanced pre-metastatic niche formation and metastasis. Mucosal Immunology, 12(4): 945-957.
DOI:10.1038/s41385-019-0144-6 |
Raux E, Lanois A, Warren M J, Rambach A, Thermes C. 1998. Cobalamin (vitamin B12) biosynthesis: identification and characterization of a Bacillus megaterium cobI operon. Biochemical Journal, 335(1): 159-166.
DOI:10.1042/bj3350159 |
Roth J R, Lawrence J G, Rubenfield M, Kieffer-Higgins S, Church G M. 1993. Characterization of the cobalamin (vitamin B12) biosynthetic genes of Salmonella typhimurium. Journal of Bacteriology, 175(11): 3 303-3 316.
DOI:10.1128/jb.175.11.3303-3316.1993 |
Shen M L, Yang R, Luo Q J, Wang S G, Ren J R. 2013. Microbial diversity of Pyropia haitanensis phycosphere during cultivation. Acta Microbiologica Sinica, 53(10): 1 087-1 102.
(in Chinese with English abstract) |
Shi W C, Li M C, Wei G S, Tian R M, Li C P, Wang B, Lin R S, Shi C Y, Chi X L, Zhou B, Gao Z. 2019. The occurrence of potato common scab correlates with the community composition and function of the geocaulosphere soil microbiome. Microbiome, 7(1): 14.
DOI:10.1186/s40168-019-0629-2 |
Sudarikov K, Tyakht A, Alexeev D. 2017. Methods for the metagenomic data visualization and analysis. Current Issues in Molecular Biology, 24: 37-58.
DOI:10.21775/cimb.024.037 |
Svensson B. 1994. Protein engineering in the α-amylase family: catalytic mechanism, substrate specificity, and stability. Plant Molecular Biology, 25(2): 141-157.
DOI:10.1007/BF00023233 |
Theodoulou F L, Kerr I D. 2015. ABC transporter research: going strong 40 years on. Biochemical Society Transactions, 43(5): 1 033-1 040.
DOI:10.1042/BST20150139 |
Uritskiy G V, DiRuggiero J, Taylor J. 2018. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome, 6(1): 158.
DOI:10.1186/s40168-018-0541-1 |
Warren M J, Raux E, Schubert H L, Escalante-Semerena J C. 2002. The biosynthesis of adenosylcobalamin (vitamin B12). Natural Product Reports, 19(4): 390-412.
DOI:10.1039/b108967f |
Watanabe F. 2007. Vitamin B12 sources and bioavailability. Experimental Biology and Medicine, 232(10): 1 266-1 274.
DOI:10.3181/0703-MR-67 |
Wichard T, Beemelmanns C. 2018. Role of chemical mediators in aquatic interactions across the prokaryote-eukaryote boundary. Journal of Chemical Ecology, 44(11): 1 008-1 021.
DOI:10.1007/s10886-018-1004-7 |
Xu H B, Luo X, Qian J, Pang X H, Song J Y, Qian J R, Chen J H, Chen S L. 2012. FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One, 7(12): e52249.
DOI:10.1371/journal.pone.0052249 |
Xu Y, Huang L, Ji D H, Chen C S, Zheng H K, Xie C T. 2015. Construction of a dense genetic linkage map and mapping quantitative trait loci for economic traits of a doubled haploid population of Pyropia haitanensis (Bangiales, Rhodophyta). BMC Plant Biology, 15(1): 228.
DOI:10.1186/s12870-015-0604-4 |
Yan Y W, Yang H C, Tang L, Li J, Mao Y X, Mo Z L. 2019. Compositional shifts of bacterial communities associated with Pyropia yezoensis and surrounding seawater co-occurring with red rot disease. Frontiers in Microbiology, 10: 1666.
DOI:10.3389/fmicb.2019.01666 |
Yang R, Fang W Y, Shan Y Y, Chen H M, Sun X, Ye Y F. 2008. Genetic diversity of epiphytic bacteria in Porphyra yezoensis. Acta Oceanologica Sinica, 30(4): 161-168.
(in Chinese with English abstract) DOI:10.3321/j.issn:0253-4193.2008.04.020 |
Yurkov V, Jappe J, Vermeglio A. 1996. Tellurite resistance and reduction by obligately aerobic photosynthetic bacteria. Applied and Environmental Microbiology, 62(11): 4 195-4 198.
DOI:10.1128/aem.62.11.4195-4198.1996 |
Zhang H, Yohe T, Huang L, Entwistle S, Wu P Z, Yang Z L, Busk P K, Xu Y B. 2018. DbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Research, 46(W1): W95-W101.
DOI:10.1093/nar/gky418 |